Adult Vitamin D Deficiency Impairs Hippocampal-Dependent Memory and Proteins Related to Synaptic Plasticity and Metabolism in Mice
Abstract
Vitamin D deficiency has been associated with a range of neuropsychiatric and neurodegenerative disorders, including schizophrenia, autism, depression, Alzheimer's disease and cognitive impairment. The active form of vitamin D, 1,25(OH)2D3, is a neurosteroid that has a wide range of functions within the adult brain including calcium maintenance, regulation of neurotrophic factors, neurotransmission, and neuroprotection. Using a model of adult vitamin D (AVD) deficiency in BALB/c mice, we have previously shown an imbalance between excitatory and inhibitory neurotransmission, which is relevant to many neuropsychiatric disorders. The aim of this study was to determine whether AVD deficiency had an effect on hippocampal function. We first analysed the proteome of the hippocampus and found evidence of altered glutamate and GABA signaling, that was consistent with previous studies, with proteomic changes related to synaptic plasticity and glutathione synthesis. We measured the concentration of glutathione in whole brain and showed that AVD- deficient mice had reduced glutathione levels. We next used a selective hippocampal- dependent learning and memory task, active place avoidance, and found that AVD-deficient mice took longer to learn the task compared to controls. Finally, structural and diffusion tensor imaging using MRI did not reveal any gross alterations in hippocampal structure or white matter bundles within the brains of AVD-deficient mice. Taken together, these results indicate that vitamin D deficiency in otherwise healthy adult rodents is sufficient to impair cognition and alter brain function, which provides important new convergent clues for understanding how vitamin D deficiency impacts on the adult brain.
Keywords
Vitamin D, brain function, Animal model, Proteomics, DTI, Ingenuity, Glutathione
Introduction
Vitamin D is a neurosteroid that exerts its effects either as a transcription factor regulating gene expression [1] or via rapid non-genomic actions through signal transduction systems [2]. The genomic effects of vitamin D are widespread with many thousands of vitamin D response elements found genome wide [1]. There is considerable evidence to show that vitamin D has many functions in the adult brain, including calcium homeostasis and signaling, regulation of neurotrophic factors, neuroprotection, and modulating neurotransmission and synaptic plasticity [3]. Moreover, there is evidence from epidemiological studies linking vitamin D deficiency in adulthood and a range of neuropsychiatric and neurodegenerative disorders [4-6].
The evidence that vitamin D deficiency can lead to cognitive impairments is mixed, and cross- sectional studies are unable to exclude reverse causality. However, a few prospective studies link low baseline vitamin D levels with subsequent mental states, particularly later in life [7]. For example, a study in community dwelling older women showed that participants with deficient serum vitamin D levels had greater cognitive impairments on the Pfeiffer Short Portable Mental State Questionnaire, compared to participants with sufficient vitamin D levels [8]. A recent prospective study has shown that vitamin D deficiency was associated with accelerated decline in cognitive domains including episodic memory and executive function [9]. Moon [10] found that vitamin D deficient patients had lower fractional anisotropy (FA) values, a measure of white matter bundles, on MRI scans from vitamin D insufficient patients primarily in frontal regions of the brain and greater memory deficits, compared to patients with sufficient vitamin D levels. However, no causal role of vitamin D in cognitive impairment has been established.
We have previously used a model of adult vitamin D (AVD) deficiency in BALB/c mice and shown in whole brain tissue that amino acid metabolism is altered, with reduced levels of glutamate and glutamine and elevated levels of γ-aminobutyric acid (GABA) and glycine [11]. We have recently shown that AVD deficiency did not affect proliferation or incorporation of adult hippocampal neurons within the dentate gyrus, at baseline or after voluntary wheel running in BALB/c mice [11]. However, we found that levels of glutamic acid decarboxylase (GAD) 65 and GAD67, which are involved in the metabolism of glutamate and GABA, were down-regulated in the brains of AVD-deficient mice [12]. An imbalance in glutamate/GABA systems has been implicated in a range of neurological disorders including schizophrenia, autism and cognitive impairment [13-15]. One potential role for the active metabolite of vitamin D, 1,25(OH)2D3, in the brain may be through altered calcium homeostasis. For example, 1,25(OH)2D3 decreases voltage-dependent calcium current (VDCC) expression in hippocampal cultures [16] and hippocampal VDCC expression increases with age and inhibits synaptic plasticity [17]. Vitamin D has also been shown to have a role in antioxidant metabolism via down regulation of the expression of inducible nitric oxide synthesis (iNOS) [18] and regulation of γ-glutamyl transpeptidase [19], an enzyme important in the glutathione pathway. More recently, an invivo study has shown that treatment with 1,25(OH)2D3 leads to upregulation of enzymes involved in glutathione synthesis, such as glutathione reductase and glutamate cysteine ligase, and to an increase in glutathione levels [20].
A previous study in adult rats showed that eight weeks of a vitamin D depleted diet led to decreased induction of long term potentiation (LTP) in the hippocampus of anesthetized animals, compared to rats fed on a control diet or vitamin D supplemented diet [21]. LTP is important in learning and memory and it is well known that glutamate neurotransmission plays a central role in LTP [22]. The active place avoidance (APA) task is a hippocampal-dependent learning and memory task for rodents that is dependent on avoidance of an aversive event. The APA has greater complexity than the more widely used Morris water maze, and yet only requires a single trial per day [23, 24]. The APA test requires the hippocampus during both acquisition and for performance once the procedure is learnt [25], although there are few studies to date that have used APA to test the effect of AVD deficiency on hippocampal-dependent learning and memory.
The primary aim of this study was to determine whether AVD deficiency had an effect on hippocampal function. We first analysed the proteome of the hippocampus to examine the effect of AVD deficiency on altered excitatory and inhibitory neurotransmitter systems. To confirm these proteomic findings we used a number of different approaches. These included measuring the level of glutathione in whole brain and using the APA to measure hippocampal- dependent learning and memory task. Finally, we conducted structural and diffusion tensor imaging using MRI to measure brain structure and neuronal integrity. Our hypothesis was that AVD-deficient BALB/c mice would show changes in the expression of proteins specifically involved with glutamate and GABA signaling, synaptic plasticity, calcium regulation and neuroprotection and that this would be associated with structural and functional changes within the hippocampus.
Methods
Animals
Ten-week old male BALB/c mice (Animal Resources Centre, Canning Vale, WA, Australia) were obtained and housed in groups of 3-4 in individually ventilated OptiMICE cages (Animal Care Systems, CO, USA), with bedding (Sanichips, Harlan Laboratories, USA) and nesting material at the Queensland Brain Institute Animal House Facility, University of Queensland.
Mice were assigned to either a control diet (Standard AIN93G Rodent diet with 1,500 IU vitamin D3/kg (prior to irradiation with 25 kGy), Specialty Feeds, WA, Australia) or a vitamin D-deficient diet (Vitamin D Deficient with 0 I.U vitamin D3/kg, AIN93G Rodent diet, Specialty Feeds, WA, Australia) for 10 weeks, until they were 20-weeks old (or 30-weeks old for the LTP experiment). The mice were maintained on a 12-hour light-dark cycle (lights on at 07:00 h) with ad libitum access to food and water. They were housed under incandescent lighting free from UVB radiation. All experimental work was performed with approval from the University of Queensland Animal Ethics Committee, under the guidelines of the National Health and Medical Research Council of Australia.
Sixty-one mice were used in this study; 8 mice for were used in the proteomics analysis (n=4 control and 4 AVD-deficient mice), 23 mice were included in the APA experiment (n=11 control and 12 AVD-deficient mice), 15 mice were included for the MRI analysis (n=7 control and 8 AVD-deficient mice), and 15 mice were used to assess glutathione levels (n=8 control and 7 AVD-deficient mice).
Proteomics
Tissue collection
The mice were euthanized by CO2, followed by decapitation. The whole brain was removed and the whole hippocampus was collected using a mouse 1mm brain block (David Kopf Instruments, California, USA) and free-hand dissection using The Mouse Brain in Stereotaxic Coordinates as a reference [26]. Tissue was frozen on dry ice and stored at -80℃ until further processing.
Tissue preparation
Hippocampal tissue from two of four mice from each cage, totaling 8 mice (n = 4 control and 4 AVD-deficient) was used in the proteomics analysis. The hippocampal tissue was homogenized and then sonicated in 8M urea, 20mM Tris, 4% Chaps and 1% DTT lysis buffer with 1X protease and phosphatase inhibitors (Roche Diagnostics, Castle Hill, AUS). The samples were placed on a rotator and spun slowly for 4 h, followed by centrifugation at 15℃ for 30 min at 13,000 g. The supernatant was removed and placed into a new tube. Protein concentration of the supernatant was determined using a Bradford assay following the standard protocol.
iTRAQ labeling
For each sample, 100 μg of protein was reduced with 2.5 mM TCEP (60ºC for 1 h and cysteine residues alkylated with 10 mM MMTS (room temperature for 15 min). Protein was precipitated with 4 volumes of ice-cold acetone overnight at -20℃. Protein was collected by centrifugation and the pellet resuspended in 0.5 M TEAB containing 0.1 % SDS. Protein was digested with sequencing grade trypsin (Promega, WI, USA) overnight. For each sample, the isobaric tagging iTRAQ reagent (AB Sciex, CA, USA) was added to the digest and incubated for 1 h at room temperature. After labeling the eight-labeled peptide samples were pooled and evaporated to dryness in a speed vac.
SCX Peptide Fractionation
The labeled peptide mix was resuspended in 0.1 % acetic acid containing 2% acetonitrile. Chromatography was performed on an 1100 series HPLC (Agilent, CA, USA) equipped with a fraction collector. Samples were fractionated using a Zorbax 300-SCX column (5 μm, 4.6 x 50 mm, Agilent, CA, USA) using a linear gradient from 0-250 mM ammonium acetate containing 2 % acetonitrile at 0.5 mL per minute with 30 s fractions. Fractions were combined accordingly to give a total of 15 fractions. Combined fractions were desalted using Empore C- 18 7mm/ 3 mL SPE cartridges (Agilent, CA, USA).
Mass Spectrometry
Peptides were dissolved in 1% formic acid and injected onto a Shimadzu Prominence nanoLC system (Shimadzu, Kyoto, Japan) at a flow rate of 1 μL/min onto a Vydac Everest C18 300 A, 5 μm, 150 mm x 150 μm column (Grace, Colombia, MD, USA). Chromatographic separation was performed using a linear gradient 10-60% solvent B over 48 min. Mobile phases consisted of solvent A (1% acetonitrile, 0.1% formic acid) and solvent B (0.1% formic acid, 90% acetonitrile). The HPLC eluent was interfaced to a TripleTOF 5600 LC/MS/MS system using a Nanospray III ionisation source (Applied Biosystems, Forster City, CA). Source conditions included an ion spray voltage of 2700 V, nebulizer gas flow of 10, curtain gas flow of 30, interface heater temperature at 150℃ and collision-induced dissociation settings included CAD gas of 10 and a declustering potential of 80 V. MS TOF spectra were acquired across the range m/z 350 - 1800 for 0.5 s followed by 20 data-dependent MS/MS measurements on pre-cursor ions (100 counts/s threshold, +2 to +5 charge state, collision energy of 40 with collision energy spread of 15). Data was acquired and processed using Analyst TF 1.5™ software (AB Sciex, CA, USA).
Protein identification and quantification
ProteinPilot™ v4.5 software (Applied Biosystems, Forster City CA) utilising the Paragon Algorithm was used for the identification of proteins. MS/MS data was searched against the Uniprot FASTA formatted database. Search parameters included trypsin as the digestion enzyme, MMTS as cys-modification, biological modifications allowed (using the set provided with the software) and 'thorough' search setting. The criterion for a positive identification was a confidence score of ≥ 95% and a minimum of 2 peptides per protein. Proteins were considered significantly different between control and AVD-deficient diets when a p-value of less than 0.05 was given. Without correcting for multiple comparisons, a dataset of these proteins was generated with fold change from control and the UniProtKB protein codes for analysis using Ingenuity Pathway Analysis (IPA) (Ingenuity System Inc, Redwood City, CA).
Ingenuity pathway analysis
Data was analysed using the 'Core Analysis' function in IPA. Data was analysed in the context of canonical pathways, biological processes and networks. The proteins included in the analysis were not corrected for multiple comparisons, so that a larger list of candidate proteins (the dataset) could be analysed using IPA. However, results from the pathway analysis were only considered significant with p-values less than 0.01. Significance of the canonical pathways was tested by the Fisher Exact test p-value. The significance calculated was a measurement of the likelihood that the pathway is associated with the dysregulated proteins by chance. Canonical pathways were ordered by significance and the ratio value (number of significant proteins in a given pathway, divided by total number of molecules that make up that pathway). Biological functions were grouped in: Diseases and Disorders, Molecular and Cellular Functions and Physiological System Development and Function. Proteins from the dataset that were associated with bio functions in the Ingenuity Pathways Knowledge Base were considered for analysis. Fischer's exact test was used to calculate a p-value determining the probability that each biological function assigned to the dataset was due to chance alone.
HPLC analysis
Mice were euthanized by CO2, followed by decapitation. The whole brain was removed and the left hand cerebrum was collected using free-hand dissection. Tissue was frozen on dry ice and stored at -80℃ until further processing for HPLC analysis.
Tissue extracts were analysed for total glutathione based on the method described by [27]. Basically, analyses were performed on an Agilent 1100 series HPLC system equipped with a Coulochem III electrochemical detector with a glassy carbon electrode (ESA Inc., MA, USA). Analyte separation was performed on a Jupiter C18 5 μm 300A column (4.6 x 250 mm, Phenomenex, NSW, Australia) using an isocratic mobile phase consisting of 3 % methanol and 0.1 % TFA in water. The column flow rate was 0.7 ml/min and analytes were detected using an electrochemical detector in DC mode, using a detector potential of 900 mV and 1000 mV potential for the guard cell. The data was acquired and processed using Chemstation software (Agilent, CA, USA).
Active place avoidance
The apparatus for the APA (Bio-Signal Group, NY, USA) consisted of an elevated arena with a grid floor surrounded by a 30 cm high transparent circular barrier, producing an enclosed arena 77 cm in diameter. The arena was located in a room with visual cues placed on the walls. The arena, including the grid floor, rotated clockwise at a speed of 1 rpm and an electric shock could be delivered through the grid floor. The position of the animal in the arena was tracked using an overhead camera linked to Tracker software (Bio-Signal Group, NY, USA). During trials, a mouse was placed in the arena and trained to avoid a 60° shock zone, the positioning of which was kept constant in relation to the room and to the external visual cues; the mouse's "start" position was always opposite the shock zone, near the barrier of the arena.
On Day 1 (habituation day), the mice were free to explore the arena with no shock given, for 5 min. On the next 4 consecutive days, Days 2 to 5 (test days), entrance into the shock zone led to the delivery of a brief foot shock (500 ms, 60 Hz, 0.5 mA). If, after the initial shock, the animal remained in the shock zone, further shocks were delivered at 1.5 s intervals until the animal moved out of the zone. Each test session lasted 10 min and recorded tracks were analyzed offline using Track Analysis software (Bio-Signal Group, NY, USA). To assess the extent of learning, the following parameters were measured: number of foot shocks delivered during the 10 min trial, latency to first entry into the shock zone, and the distance travelled during the trial.
The second APA test that was performed at 20-weeks old, had the location of the shock zone and start zones moved, compared to the initial APA at 9-weeks old, to increase the difficulty of the task. The extra-maze cues were also changed for the second APA test.
Magnetic resonance imaging
Mice were anaesthetized by an i.p injection of Lethabarb at 100 mg/kg body weight and transcardially perfused with 40 ml of phosphate buffered saline (PBS) and 40 ml of 4 % paraformaldehyde (PFA) in 0.1 M PBS. The brains were removed and post fixed in 4 % PFA for 24 h before being stored in sodium azide (0.05%) in 0.1M PBS (PBS azide) for future analysis.
Brains were incubated in 0.1% Magnevist® (gadopentetate dimeglumine, Bayer Healthcare Pharmaceuticals Inc., NJ, USA) in PBS for 4 days prior to imaging. MRI data were acquired with the samples immersed in Fomblin fluid (Solvay Solexis, Italy), using a 16.4 T (89 mm) Bruker micro-imaging system (Bruker Biospin, Karlsruhe, Germany) and a 15 mm SAW coil (M2M Imaging, USA). High-resolution anatomical images were acquired using three- dimensional (3D) gradient-echo FLASH (fast low angle shot) T1/T2*-weighted using repetition time (TR) 50 ms, echo time (TE) 12 ms, number of excitation averaging (NEX) 1 at 30 micron isotropic resolution. 3D Diffusion-weighted images (DWI) were acquired using Stjskal-Tanner DW spin-echo sequence using the following parameters: TR = 400 ms; TE = 22.8 ms; 80 micron isotropic resolution and a signal average of 1. DWI data were composed of two images acquired without diffusion weighting (b0) and thirty direction DW images (b value of 5000 s/mm2, δ/Δ = 2.5/14 ms). The samples were maintained at 22℃.
Brain volumetric measurements using segmented 3D MRI atlas
Anatomical MR images were cleaned of any non-brain tissue using a mask created in ITK- SNAP (www.itksnap.org). Inter-group differences in brain volumes were mapped using voxel-based morphometry [28]. Firstly, a study-specific BALB/c template was created using Advanced Normalization Tools (ANTs) buildtemplateparallel script (http://picsl.upenn.edu/software/ants/), where high-resolution gradient-echo images of all mice from the study (N = 15) were aligned to a common reference space via a 12 degrees-of-freedom (DOF) affine alignment, followed by 5 iterative symmetric diffeomorphic registrations [29]. Secondly, the resulting BALB/c template was registered using ANTs diffeomorphic registration to an adult C57Bl/6 MRI atlas that contains 18 segmentations of brain structures [30]. Finally, the warp information from the previous steps was used to register the segmented structures back to the individual subjects for analysis of inter-group differences in brain region volumes. Volumes for each brain segment were calculated using ITK-SNAP. See Table 1 for brain regions analysed.
Voxel based morphometry of DTI maps
Non-brain tissue was removed from diffusion MR images by creating a mask of each brain in ITK-SNAP. DTI maps, consisting of fractional anisotropy (FA), mean diffusivity (MD), axial diffusivity (AD) and radial diffusivity (RD), were generated using MRTrix 0.2.9 (www.mrtrix.org). A study specific FA template was created using ANTs build template parallel script, and subsequently the other of DTI parameters were co-registered to the template space. VBM analyses were performed using FSL randomise (fsl.fmrib.ox.ac.uk) using 5000 permutations [31], threshold-free cluster enhancement and correction for multiple comparisons. The FA, MD, AD, and RD maps were assessed for inter-group differences using tract-based spatial statistics (TBSS) as previously described [32]. All FA images were aligned onto the ANTs FA template using a nonlinear registration algorithm within the TBSS package and the white matter tracts were skeletonized (using FA threshold of 0.3) to create a skeleton template. Subsequently, all DTI maps from each subject were skeletonized. The FA skeleton from each sample was registered to the template using TBSS nonlinear registration method.
The other DTI maps (MD, AD, and RD skeletons) are co-registered to the template by applying the same transformation calculated for the FA. TBSS analyses were performed using 5000 permutations (31), threshold-free cluster enhancement and correction for multiple comparisons. The resulting t-map was thresholded > 0.95 to assess statistical significance.
Statistical Analysis
Results were analysed for statistical significance using SPSS (version 20.0) software. All data were analysed for the main effects of Diet (control or AVD-deficient) using analysis of variance (ANOVA) or, where appropriate, repeated measures ANOVA. Significant differences (p < 0.05) were followed up with post-hoc t-tests.
Results
AVD deficiency altered the hippocampal proteome
Using iTRAQ proteomics we identified 2,648 proteins, 1,373 of these proteins had 2 or more peptides identified with > 95% confidence. All other proteins had less peptides (0 or 1) identified with 95% confidence. Levels of 45 proteins were significantly different between control and AVD-deficient mice (p < 0.05) and 32 of these had a fold change >1.5. Of the significantly different proteins, 15 proteins were up-regulated following AVD deficiency and 30 proteins were down-regulated (Table 1). For all proteins identified using protein pilot and their quantification values, see Supplementary File 1.
As hypothesized a number of proteins were identified that were significantly different with AVD deficiency that are involved in glutamate and GABA signaling; including glutamine synthetase, neutral amino acid transporter A, GABA transporter 1 and glutamate decarboxylase 2. Furthermore, we identified an effect of AVD deficiency on an R-type calcium channel subunit and two proteins involved in glutathione synthesis, including glutathione synthetase and 5-oxoprolinase.
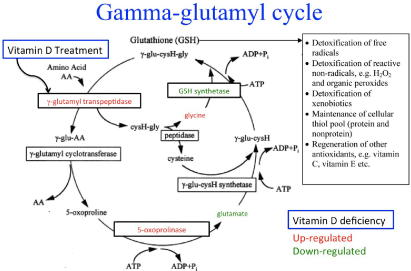
Analysis using Ingenuity Pathway Analysis (IPA) showed the top canonical pathway affected by AVD deficiency was the γ-glutamyl cycle (p = 0.0004) (Table 2), which is involved with the synthesis of glutathione, biosynthesis of reductants and may be involved with amino acid transport [33]. Other canonical pathways that were identified as significantly different were Glutamine Biosynthesis I (p = 0.002), Glutamate Receptor Signaling (p = 0.006), Glutathione Biosynthesis (p = 0.006) and GABA Receptor Signaling (p = 0.008). See Supplementary File 2 for the full list of significant (p < 0.01) canonical pathways.
The top six most significant biological functions associated with the dataset were 'Small Molecule Biochemistry' (p < 0.001, 19 molecules), 'Cell-To-Cell Signaling and Interaction' (p < 0.001, 17 molecules), 'Molecular Transport' (p < 0.0001, 17 molecules), 'Cellular Assembly and Organisation' (p < 0.001, 15 molecules), 'Amino Acid Metabolism (p < 0.001, 10 molecules) and 'Behaviour' (p < 0.001, 10 molecules). These bio functions are listed in Table 3.
Within these significant biological functions, IPA identified a number of processes associated with the dysregulated proteins including release of amino acids and neurotransmitters, uptake and metabolism of amino acids, plasticity of synapse, quantity of vesicles and synapses, retraction of dendrites and remodeling of actin filaments; as well as behaviours involved in emotion, contextual conditioning, fear, and learning and memory. To view all biological processes identified by IPA grouped by main biological function and then in order of significance see Supplementary File 3.
AVD deficiency was associated with reduced whole brain glutathione levels
In conjunction with the two proteins involved in the γ-glutamyl cycle that were differentially regulated with AVD deficiency we examined whether the synthesis of glutathione was altered.
There was a significant effect of Diet (F1,13 = 26.43, p < 0.001) on total glutathione levels, with a significant decrease in the levels of glutathione in AVD-deficient mice (mean ± SEM; 513.27 μg/mg ± 53.72 for controls and 210.91 μg/mg ± 12.22 for AVD-deficient mice).
Hippocampal-dependent learning and memory was impaired in AVD-deficient mice
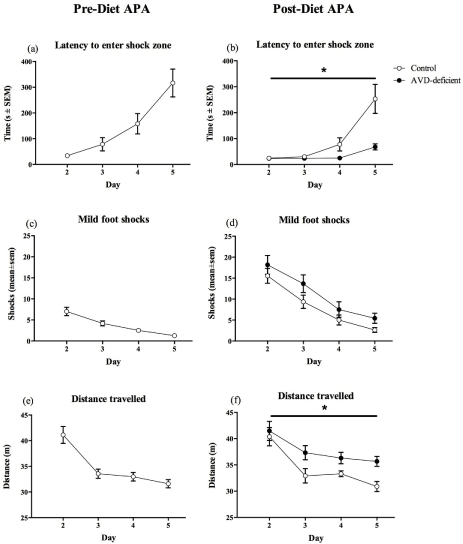
Mice were tested prior to dietary manipulation to gain a baseline result in the learning and memory task and to ensure that there was no difference between the two groups. All mice learnt to avoid the shock zone over the four test days (F1,22 = 34.40, p < 0.001, Figure 1.c) and their latency to enter the shock zone was significantly increased over the four days of testing (F1,22= 13.08, p < 0.001, Figure 1.a). The distance travelled during the 10 min test was significantly reduced over the course of the four test days for all mice (F1,22 = 29.29, p < 0.001, Figure 1.e). These results showed that all mice were able to learn the paradigm.
Following ten weeks of dietary manipulation the mice were retested. During this second round of testing, there was a significant effect of Diet (F1,21 = 15.52, p = 0.001, Figure 1.b) on the latency to enter the shock zone over the test days, with controls showing an increased latency compared to AVD-deficient mice. There was no significant effect of Diet (F1,21 = 0.60, p = 0.446) on the maximum time to avoid shock over the course of the test days but there was a significant effect of Diet (F1,21 = 5.30, p = 0.032) on Day 2, with a shorter maximum time to avoid in the AVD-deficient mice (209.18 s ± 31.93 in Controls and 124.67 s ± 19.52 in AVD-deficient mice).
The number of shocks received by all mice was significantly reduced over the test days (F1,21= 47.49, p < 0.000). Furthermore, there was no significant effect of Diet (F1,21 = 2.62, p = 0.121, Figure 1.d) on the number of shocks over the four test days. On the final test day, Day 5, the number of shocks was no longer significant (F1,21 = 4.10, p = 0.056). However, when analyzing the first 5 min of Day 5, AVD-deficient mice received significantly more shocks compared to controls (F1,21 = 6.27, p = 0.021) (1.09 shocks ± 0.28 in controls and 3.83 shocks ± 1.01 in AVD-deficient mice), but not in the second 5 min (F1,21 = 0.01, p = 0.947) of Day 5 (1.55 shocks ± 0.41 in controls and 1.58 shocks ± 0.38 in AVD-deficient mice). Furthermore, there was no significant difference in the number of shocks between the first 5 min and the second 5 min by control mice (F1,10 = 1.10, p = 0.320). However, there was a significant reduction in shock number for AVD-deficient mice in the second 5 min (F1,11 = 5.75, p = 0.035), suggesting that they were still learning how to avoid the shock zone.
There was no significant main effect of Diet (F1,21 = 2.95, p = 0.180) on the distance travelled over the five days of testing. There was however, a significant Diet x Day interaction (F1,21 = 3.85, p = 0.015, Figure 1.f), with AVD-deficient mice exhibiting hyperlocomotion compared to controls towards the end of the testing. On Day 1, there was no significant effect of Diet (F1,21 = 1.89, p = 0.184) on the distance travelled during habituation to the arena. On test days, when shock was applied to the shock zone, there was still no effect of Diet (F1,21 = 0.19, p = 0.667) on the distance travelled on Day 2. However, there was a significant effect of Diet (F1,21= 5.55, p = 0.028) over the four test days, with AVD-deficient mice travelling more than the controls.
Assessing the difference between the number of shocks received on the final day of testing before being placed on diet, compared to the number of shocks on the final day of testing after dietary manipulation, there was a significant Diet x Day (F1,21 = 5.14, p = 0.034) effect. AVD- deficient mice received significantly more shocks during the second APA test compared to control mice.
AVD deficiency was not associated with changes in brain structure or neuronal integrity
Magnetic resonance imaging was used to assess gross brain structure, and neuronal integrity via diffusion tensor imaging (DTI). There was no significant effect of Diet (F1,13 = 0.16, p = 0.698) on total brain volumes (Controls = 387.35 ± 3.90 mm3 and AVD-deficient = 383.69 ± 7.90 mm3). Total brain volumes excluded the olfactory bulbs, which are sometimes damaged during brain extraction. There was also no significant effect of Diet on any white matter structure volume (F1,13 < 2.20, p > 0.160) or brain region volume, including the hippocampus (F1,13 < 1.49, p > 0.244). See Table 4 for details. Using both voxel based morphometry (VBM) and tract based spatial statistics (TBSS) analysis of the FA, median diffusivity (MD), axial diffusivity, and radial diffusivity maps showed no significant differences between control and AVD-deficient mice throughout the brain following threshold-free cluster enhancement and correction for multiple comparisons. Mean whole brain values were also analysed for FA (0.225 ± 0.005 for controls and 0.216 ± 0.002 for AVD-deficient mice, F1,13 = 0.47, p = 0.505) and MD (0.301 ± 0.003 for controls and 0.297 ± 0.003 for AVD-deficient mice, F1,13 = 1.03, p = 0.330) and there were no significant differences seen between control and AVD-deficient mice. AVD deficiency did not alter neuronal integrity following 10 weeks of dietary manipulation in adult male BALB/c mice.
Discussion
The main findings from this study indicate convergent evidence of an effect of AVD deficiency on glutamate and GABA signaling, synaptic plasticity and on glutathione synthesis that is associated with impaired hippocampal-dependent learning and memory. Taken together, these results provide further evidence that vitamin D deficiency in otherwise healthy adult rodents is sufficient to impair cognition and alter brain function. We identified a number of canonical pathways affected by AVD deficiency including the γ-glutamyl cycle, important for the production of glutathione, an important antioxidant; and glutamate and GABA receptor signaling and glutamine biosynthesis. Furthermore, we identified some key biological functions that may be affected by AVD deficiency including small molecule biochemistry, cell-to-cell signaling and interaction, molecular transport, cellular assembly and organization and amino acid metabolism. We did not see changes in white matter bundles, as measured be DTI, suggesting that hippocampal alterations are an early consequence of vitamin D deficiency.
The proteomic analysis also identified decreased hippocampal expression of CaV2.3 in AVD- deficient mice. This protein makes up the α1E subunit of the R-type voltage-dependent calcium channel (VDCC). CaV2.3 is expressed in both pre-and post-synaptic structures of mouse hippocampus, with strong expression in the stratum radiatum [34]. It is still not clear what role R-type VDCCs play in synaptic processing in the hippocampus. Dendritic spine calcium influx is inhibited by R-type VDCCs, apparently via selective activation of a Ca-activated K+- channels [35]. Alternatively, R-type VDCCs of the dendritic tufts enhance plateau potentials in CA1 neurons [36]. In addition, pre-synaptic R-type calcium channels increase glutamate release in the hippocampus, amplifying post-synaptic depolarisation [37]. Calcium influx is an early molecular requirement for LTP induction [38]. R-type VDCC inhibition prevents coincident perforant pathway and Schaffer collateral LTP induction [36] and CaV2.3 has been implicated in calcium influx during presynaptic LTP induction [39]. Of relevance to the present study is that Cav2.3 knockout mice exhibit impaired spatial memory [40].
We have previously shown that AVD deficiency in male mice leads to reduced glutamate levels and increased GABA levels, measured by HPLC in whole brain; and a small but significant reduction in the GAD enzymes in whole brain, measured by western blot [12]. We have now provided further evidence to show that there is a significant reduction in GAD65 in the hippocampus of male BALB/c mice using proteomic analysis.
Studies in knockout mice have shown that ablation of GAD67 results in neonatal lethality and reductions in GABA levels to 7% of WT mice [41]. GABA is necessary during development to regulate neocortical neurogenesis [42]. However, in GAD65 knockout mice, there was no change to GABA levels and it was shown that GAD65 is not required for development but is important for modulating inhibitory neurotransmission in response to increased demand [43]. GAD65 is localized to axon terminals and is reversibly bound to the membrane of synaptic vesicles [44, 45]. Further studies in the GAD65 knockout mice have shown that GAD65 mediates activity-dependent GABA synthesis [46] but moreover, has a significant impact on GABA release during sustained activation, possibly through mobilization of vesicles or replenishment of vesicles at the synapse [47]. Therefore, it is feasible that the reduction in GAD65 protein expression associated with AVD-deficiency could have an impact on GABAergic inhibitory neurotransmission.
Another important protein involved in GABA signalling that was found to be upregulated by AVD deficiency was the GABA transporter 1 (GAT1 or SLC6A1). GAT1 is also localized to GABAergic interneuron axons and nerve terminals and is responsible for GABA reuptake from the synaptic cleft [48]. With reductions in GABA synthesis, due to lower levels of the GAD65 enzyme, it is plausible to suggest that this would lead to upregulation of GAT1 in an attempt to restore intracellular GABA levels. Reductions in GAD65 in a pathophysiological pain model, did accompany elevated levels of GAT1, although it was unclear which protein expression level change preceded the other [49].
AVD deficiency and oxidative stress
The top recognized pathway altered by AVD-deficiency was the γ-glutamyl cycle with significant changes in the level of two enzymes; glutathione synthetase was down-regulated and 5-oxoprolinase was up-regulated as shown in Figure 2. A secondary significant canonical pathway was also identified involving glutathione, which was glutathione biosynthesis. We have previously published a reduction in glutamate levels and an increase in glycine levels with AVD deficiency in whole brain [12] and these changes have also been added to Figure 2.
Furthermore, vitamin D treatment has previously been shown to up-regulate γ-glutamyl transpeptidase and increase glutathione levels invitro (19) (also noted on Figure 2). 5- oxoprolinase may be up-regulated in an attempt to compensate for lower glutamate levels and with a reduction in glutamate plus a reduction in the level of glutathione synthetase, this could lead to the lower levels of glutathione seen in AVD-deficient mice.
Using both proteomics and HPLC, we have shown that the γ-glutamyl cycle has been dysregulated following AVD deficiency leading to a reduction in total glutathione levels within the brain. Oxidative stress can lead to cell membrane dysfunction, DNA damage and inactivation of proteins, eventually leading to cell death. Studies have shown that people with mild cognitive impairment have higher levels of oxidative stress compared to healthy controls [50]. Furthermore, a study performed in rats showed that even in young animals (3 months of age), induction of oxidative stress led to impairments in spatial learning and memory [51]. Induced oxidative stress, via exposure to 100% oxygen for 48 h, produced an abnormal accumulation of synaptic vesicles in swollen nerve terminals and a reduction in neurotransmission [52]. In addition, following the induction of oxidative stress, synaptic membranes were shown to have elevated markers of oxidative stress including lipid peroxides and protein carbonyls, and a change in membrane surface potential. It was proposed that this would result in reduced membrane fusion between nerve terminal membranes and synaptic vesicles and produce the subsequent decline in neurotransmission shown previously [51, 52].
Synaptic plasticity
Analysing the differentiated proteins from the proteomics using IPA provided us with interesting candidate pathways that may underpin the changes found in hippocampal- dependent learning and memory in AVD deficiency. The analysis revealed that a number of proteins down regulated with AVD deficiency were involved in synapse formation, vesicle formation and release, synapse strength and plasticity. Reductions in APP, PPFIA3, MECP2 and RASGRF1 have all previously been reported to impair synaptic plasticity and were all down regulated in AVD-deficient mice [53-56].
RASGRF1 normally facilitates neurite outgrowth in response to NGF and BDNF (54), yet is decreased with AVD deficiency. RASGRF1 knockout mice have impaired long-term memory but intact learning and short-term memory in an emotional conditioning task [57]. In addition, hippocampal-dependent learning using the Morris water maze was impaired in these mice [58]. MECP2 loss-of-function mutant mice have impaired spatial learning and memory on the Morris water maze, impaired contextual conditioned fear memory and show deficits in hippocampal-dependent long-term social memory [53]. In addition, they have abnormal synaptic structure and function [53]. Although these studies show deficits with the complete absence of the protein of interest, they do provide evidence that these proteins are important for proper synaptic plasticity and favourable learning and memory. AVD-deficient BALB/c mice have not been tested on the Morris water maze, however we used an alternative hippocampal-dependent learning and memory task, the APA. There are limited numbers of studies using the APA to assess GABAergic neurotransmission on hippocampal-dependent learning and memory, although, one study using an agonist of the GABAB receptor, showed disruption of spatial avoidance using this test [59]. Studies using the active place avoidance have been used to show impairments in glutamatergic neurotransmission with systemic administration of MK-801, a non-competitive NMDA receptor antagonist. MK-801 treatment leads to increased travel on the arena, increased errors and reduced time avoiding the shock zone in Long-Evans rats [60].
Gross brain structure and neuronal integrity were not altered with AVD deficiency
Although there is significant epidemiological evidence on the association between vitamin D deficiency and cognitive impairment, dementia, Alzheimer's disease and cerebrovascular disease [8, 61, 62], very few studies have looked at the relationship between vitamin D deficiency and MRI pathologies in humans [10, 62]. For example, one study performed in humans, which was the first of its kind, showed that vitamin D deficient patients with memory complaints had lower FA values in most areas of the brain [10]. Using both VBM and TBSS, in the current study there were no differences between control and AVD-deficient mice on a range of standard DTI parameters including FA, a measure of microstructural integrity [63].
Limitations and Strengths
Using proteomics the effects of AVD deficiency on protein expression in the hippocampus was analysed and a number of candidate proteins were identified, however this was limited to one brain region, ideally future experiments should include analysis of other brain regions. Furthermore, the results from the proteomics analysis are preliminary and require further validation, via technical or proteomic replication. The proteomic results have provided a list of up and down-regulated proteins. However, this study cannot tell if these proteins are altered by vitamin D directly, altered through down-stream effects of vitamin D-targeted gene changes, or as a result of compensatory effects. To enable us to unravel the mechanism by which AVD deficiency impacts on the brain, it is important to differentiate between the two forms of protein expression changes. However, iTRAQ has been shown to be much more sensitive than both 2D difference gel electrophoresis and cleavable isotope-coded affinity tags, identifying significantly more peptides from each protein and a greater number of differentiated proteins without affecting accuracy [64].
Future Directions
This study has shown that multiple pathways involving glutathione, glutamate and GABA were dysregulated by AVD deficiency, including glutathione biosynthesis, glutamate and GABA receptor signaling, and glutamine biosynthesis. Furthermore, IPA has implicated AVD deficiency in changes to neurotransmission and amino acid metabolism. Changes to amino acid metabolism that alter the availability of glutamate and glutamine could then have downstream effects on neurotransmission and glutathione maintenance. Alternatively, elevated oxidative stress could deplete glutathione stores, leading to reductions in glutathione components, such as glutamate, which would then lead to impaired neurotransmission. Future experiments should address these issues. For example, if oxidative stress is driving all these changes, treating AVD- deficient mice with n-acetylcysteine, a molecule known to increase glutathione levels [65], should correct reductions in glutamate levels and changes in neurotransmission.
Future experiments will be needed to discover the mechanism behind AVD deficiency's impact on hippocampal-dependent learning and memory. Future studies should examine the effects of an extended period on vitamin D deficiency prior to MRI or alternatively, since most of the studies linking vitamin D deficiency to cognitive impairment, Alzheimer's disease and MRI pathology have occurred in elderly populations, a future experiment should test the effects of AVD deficiency in older mice and assess both cognitive domains and connectivity in the brain using DTI.
Importantly, the current findings provide convergent evidence to support the epidemiological link between adult vitamin D deficiency and neuropsychiatric and neurodegenerative disorders, including cognitive impairment; and has provided essential clues regarding the mechanism of how vitamin D deficiency may lead to such adverse outcomes.
Availability of supporting data
The data sets supporting the results of this article are included within the article and its additional files.
Competing interests
The authors declare no conflict of interest.
Author's contributions
NG participated in the design of the experiment, carried out the dietary manipulation, tissue collection, participated in the preparation and running of the samples, performed the statistical analysis and the pathway analysis and drafted the manuscript. PJ carried out the design, tissue preparation and performed the running of the samples for the proteomics; and performed the identification and quantification of the proteins. NK participated in the MRI experiments. JM helped to draft the manuscript. TB participated in the design and coordination of the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Suzy Alexander for assistance with the experiments and Dr. Amanda Nouwens, who is the Mass Spectrometry facility manager, School of Chemistry and Molecular Biosciences, University of Queensland.
Funding
This work was supported by the National Health and Medical Research Council of Australia grant APP1070081.
References
- Ramagopalan SV, Heger A, Berlanga AJ, et al. (2010) A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res 20: 1352-1360.
- Falkenstein E, Tillmann HC, Christ M, et al. (2000) Multiple actions of steroid hormones--a focus on rapid, nongenomic effects. Pharmacol Rev 52: 513-556.
- Groves NJ, McGrath JJ, Burne TH (2014) Vitamin D as a neurosteroid affecting the developing and adult brain. Annual review of nutrition 34: 117-141.
- Anglin RE, Samaan Z, Walter SD, et al. (2013) Vitamin D deficiency and depression in adults: systematic review and meta-analysis. Br J Psychiatry 202: 100-107.
- Annweiler C, Llewellyn DJ, Beauchet O (2013) Low serum vitamin D concentrations in Alzheimer's disease: a systematic review and meta-analysis. J Alzheimers Dis 33: 659-674.
- Balion C, Griffith LE, Strifler L, et al. (2012) Vitamin D, cognition, and dementia: a systematic review and meta-analysis. Neurology 79: 1397-1405.
- Etgen T, Sander D, Bickel H, et al. (2012) Vitamin D deficiency, cognitive impairment and dementia: a systematic review and meta-analysis. Dement Geriatr cogn disord 33: 297-305.
- Annweiler C, Schott AM, Allali G, et al. (2010) Association of vitamin D deficiency with cognitive impairment in older women: cross-sectional study. Neurology 74: 27-32.
- Miller JW, Harvey DJ, Beckett LA, et al. (2015) Vitamin D Status and Rates of Cognitive Decline in a Multiethnic Cohort of Older Adults. JAMA Neurol 72: 1295-1303.
- Moon Y, Moon WJ, Kwon H, et al. (2015) Vitamin D deficiency disrupts neuronal integrity in cognitively impaired patients. J Alzheimers Dis 45: 1089-1096.
- Groves NJ, Bradford D, Sullivan RK, et al. (2016) Behavioural effects of adult vitamin D deficiency in BALB/c mice are not associated with proliferation or survival of neurons in the adult hippocampus. PLoS One 11: e0152328.
- Groves NJ, Kesby JP, Eyles DW, et al. (2013) Adult vitamin D deficiency leads to behavioural and brain neurochemical alterations in C57BL/6J and BALB/c mice. Behav Brain Res 241: 120-131.
- Kehrer C, Maziashvili N, Dugladze T, et al. (2008) Altered Excitatory-Inhibitory Balance in the NMDA-Hypofunction Model of Schizophrenia. Front Mol Neurosci 1: 6.
- Lewis DA, Moghaddam B (2006) Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol 63: 1372-1376.
- Nelson ED, Kavalali ET, Monteggia LM (2006) MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol 16: 710-716.
- Brewer LD, Thibault V, Chen KC, et al. (2001) Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci 21: 98-108.
- Thibault O, Hadley R, Landfield PW (2001) Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J neurosci 21: 9744-9756.
- Garcion E, Sindji L, Montero-Menei C, et al. (1998) Expression of inducible nitric oxide synthase during rat brain inflammation: regulation by 1,25- dihydroxyvitamin D3. Glia 22: 282-294.
- Garcion E, Sindji L, Leblondel G, et al. (1999) 1,25-dihydroxyvitamin D3 regulates the synthesis of gamma-glutamyl transpeptidase and glutathione levels in rat primary astrocytes. J Neurochem 73: 859-866.
- Jain SK, Micinski D (2013) Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem Biophys Res Commun 437: 7- 11.
- Salami M, Talaei SA, Davari S, et al. (2012) Hippocampal long term potentiation in rats under different regimens of vitamin D: an in vivo study. Neurosci Lett 509: 56-59.
- Bashir ZI, Bortolotto ZA, Davies CH, et al. (1993) Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature 363: 347-350.
- Cimadevilla JM, Kaminsky Y, Fenton A, et al. (2000) Passive and active place avoidance as a tool of spatial memory research in rats. J Neurosci Methods 102: 155-164.
- Vukovic J, Borlikova GG, Ruitenberg MJ, et al. (2013) Immature doublecortin-positive hippocampal neurons are important for learning but not for remembering. J Neurosci 33: 6603-6613.
- Cimadevilla JM, Wesierska M, Fenton AA, et al. (2001) Inactivating one hippocampus impairs avoidance of a stable room-defined place during dissociation of arena cues from room cues by rotation of the arena. Proc Natl Acad Sci U S A 98: 3531-3536.
- Franklin GPaKBJ (2012) Paxinos and Franklin's the Mouse Brain in Stereotaxic Coordinates. (4th Edn) Elsevier Academic Press 360.
- Khan A, Khan MI, Iqbal Z, et al. (2011) A new HPLC method for the simultaneous determination of ascorbic acid and aminothiols in human plasma and erythrocytes using electrochemical detection. Talanta 84: 789-801.
- Ashburner J, Friston KJ (2000) Voxel-based morphometry--the methods. Neuroimage 11: 805-821.
- Avants BB, Tustison NJ, Song G, et al. (2011) A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54: 2033-2044.
- Ma Y, Hof PR, Grant SC, et al. (2005) A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience 135: 1203-1215.
- Nichols TE, Holmes AP (2002) Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp 15: 1-25.
- Smith SM, Jenkinson M, Johansen-Berg H, et al. (2006) Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage 31: 1487-1505.
- Vina JR, Palacin M, Puertes IR, et al. (1989) Role of the gamma-glutamyl cycle in the regulation of amino acid translocation. Am J Physiol 257: E916-E922.
- Parajuli LK, Nakajima C, Kulik A, et al. (2012) Quantitative regional and ultrastructural localization of the Ca(v)2.3 subunit of R-type calcium channel in mouse brain. J Neurosci 32: 13555-13567.
- Bloodgood BL, Sabatini BL (2007) Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53: 249-260.
- Takahashi H, Magee JC (2009) Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron 62: 102-111.
- Wu LG, Borst JG, Sakmann B (1998) R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc Natl Acad Sci USA 95: 4720-4725.
- Malenka RC, Lancaster B, Zucker RS (1992) Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron 9: 121-128.
- Breustedt J, Vogt KE, Miller RJ, et al. (2003) Alpha1E-containing Ca2+ channels are involved in synaptic plasticity. Proc Natl Acad Sci USA 100: 12450-12455.
- Kubota M, Murakoshi T, Saegusa H, et al. (2001) Intact LTP and fear memory but impaired spatial memory in mice lacking Ca(v)2.3 (alpha(IE)) channel. Biochem Biophys Res Commun 282: 242-248.
- Asada H, Kawamura Y, Maruyama K, et al. (1997) Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA 94: 6496-6499.
- LoTurco JJ, Owens DF, Heath MJ, et al. (1995) GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15: 1287-1298.
- Asada H, Kawamura Y, Maruyama K, et al. (1996) Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun 229: 891-895.
- Kaufman DL, Houser CR, Tobin AJ (1991) Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem 56: 720-723.
- Christgau S, Aanstoot HJ, Schierbeck H, et al. (1992) Membrane anchoring of the autoantigen GAD65 to microvesicles in pancreatic beta-cells by palmitoylation in the NH2-terminal domain. J Cell Biol 118: 309-320.
- Patel AB, de Graaf RA, Martin DL, et al. (2006) Evidence that GAD(65) mediates increased GABA synthesis during intense neuronal activity invivo. J Neurochem 97: 385-396.
- Tian N, Petersen C, Kash S, et al. (1999) The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci USA 96: 12911-12916.
- Minelli A, Brecha NC, Karschin C, et al. (1995) GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J Neurosci 15: 7734-7746.
- Ford A, Castonguay A, Cottet M, et al. (2015) Engagement of the GABA to KCC2 signaling pathway contributes to the analgesic effects of A3AR agonists in neuropathic pain. J Neurosci 35: 6057-6067.
- Pratico D, Clark CM, Liun F, et al. (2002) Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch Neurol 59: 972-976.
- Fukui K, Omoi NO, Hayasaka T, et al. (2002) Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann N Y Acad Sci 959: 275-284.
- Urano S, Asai Y, Makabe S, et al. (1997) Oxidative injury of synapse and alteration of antioxidative defense systems in rats, and its prevention by vitamin E. Eur J Biochem 245: 64-70.
- Moretti P, Levenson JM, Battaglia F, et al. (2006) Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci 26: 319-327.
- Talebian A, Robinson-Brookes K, MacDonald JI, et al. (2013) Ras guanine nucleotide releasing factor 1 (RasGrf1) enhancement of Trk receptor-mediated neurite outgrowth requires activation of both H-Ras and Rac. J Mol Neurosci 49: 38-51.
- Senechal Y, Kelly PH, Dev KK (2008) Amyloid precursor protein knockout mice show age- dependent deficits in passive avoidance learning. Behav Brain Res 186: 126-132.
- Bredt DS, Nicoll RA (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40: 361-379.
- Brambilla R, Gnesutta N, Minichiello L, et al. (1997) A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature 390: 281-286.
- Giese KP, Friedman E, Telliez JB, et al. (2001) Hippocampus-dependent learning and memory is impaired in mice lacking the Ras- guanine-nucleotide releasing factor 1 (Ras-GRF1). Neuropharmacology 41: 791- 800.
- Stuchlik A, Vales K (2009) Baclofen dose-dependently disrupts learning in a place avoidance task requiring cognitive coordination. Physiol Behav 97: 507-511.
- Stuchlik A, Vales K (2005) Systemic administration of MK-801, a non-competitive NMDA- receptor antagonist, elicits a behavioural deficit of rats in the Active Allothetic Place Avoidance (AAPA) task irrespectively of their intact spatial pretraining. Behav Brain Res 159: 163-171.
- Littlejohns TJ, Henley WE, Lang IA, et al. (2014) Vitamin D and the risk of dementia and Alzheimer disease. Neurology 83: 920-92 8.
- Buell JS, Dawson-Hughes B, Scott TM, et al. (2010) 25-Hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving home services. Neurology 74: 18-26.
- Pfefferbaum A, Sullivan EV, Hedehus M, et al. (2000) Age- related decline in brain white matter anisotropy measured with spatially corrected echo- planar diffusion tensor imaging. Magn Reson Med 44: 259-268.
- Wu WW, Wang G, Baek SJ, et al. (2006) Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel- or LC-MALDI TOF/TOF. J Proteome Res 5: 651-658.
- De Rosa SC, Zaretsky MD, Dubs JG, et al. (2000) N-acetylcysteine replenishes glutathione in HIV infection. Eur J Clin Invest 30: 915-929.
- Singh M, Gupta S, Singhal U, et al. (2013) Evaluation of the oxidative stress in chronic alcoholics. J Clin Diagn Res 7: 1568-1571.
Corresponding Author
Thomas Burne, Queensland Brain Institute, The University of Queensland, St Lucia, Qld 4076, Australia, Fax: +61 7-3346-6301, Phone: + 61 7-3346-6371.
Copyright
© 2022 Groves NJ. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.