Artemisinin-dipeptidyl Vinyl Sulfone Hybrid Inhibitors of Plasmodium Falciparum Falcipain 2 with Favorable Pharmacokinetic Profile
Abstract
In this work, we virtually design Artemisinin-Vinyl Sulfone Hybrid (AVSH) inhibitors of Falcipain2 (FP2) through Structure-Based Drug Design (SBDD) approach. From a series of 6 AVSH with known FP2 inhibitory activity, FP2-AVSHx complexes were built by in situ modification of the X-rays crystal structure of FP2 complexed with epoxysuccinate E64 (3BPF.pdb). We elaborated one descriptor FP2-AVSHx complexation QSAR models with linear correlation between the computed complexation Gibbs free energy (ΔΔGcom) and the measured. The first model is based on Molecular Mechanics Poisson-Boltzmann scheme.
a second one on Quantum Mechanics Polarization Continuum Model
Moreover a reliable bound conformation 3D-QSAR pharmacophore of FP2 inhibition derived from the QSAR model
provided inhibition structural information confirmed by the enzyme-inhibitor interaction energy
The breakdown of ΔEint into each active site residue contribution shed light on the suitable substitutions on the artemisinin moiety as well as the vinyl sulfone one able to improve FP2 active site pockets filling and helped in the design of novel Artemisinin-vinyl sulfone analogs with favorable pharmacokinetic profile. From this study, structural requirements for the design of potent hybrid antimalarials were assessed through the combined use of MM-PB, QM-PCM, PH4 and predicted ADME approaches.
Keywords
Malaria, Artemisinin combination therapies, FP2 hybrid inhibitors, Drug design, Quantum mechanics, QSAR model, Pharmacophore model, ADME properties
Introduction
The burden of malaria in 2015, despite controversies, reach 400,000 deaths worldwide, especially in Africa, a region which was home to 88% of cases and 90% of deaths with a population at risk estimated at over 300 million people consisting mostly children under five years [1]. Since the advent of Artemisinin Combinations Therapies (ACT) in 2006, no discovery outcome after 10 years suspending the survival of the population at risk at the tiny rope of any emergence of resistant strain to ACT. The situation is more than alarming since the parasite is already resistant to medicines combined to artemisinin in the ACT and the worse is the cases of resistance to artemisinin reported in Southeast Asia [2]. Unfortunately the deep concern is that the continent the most affected by malaria is the one in which the research effort is the lowest, making sure that reaching the critical mass of African researchers is unavoidable in the hope of overcoming this disaster one day.
The rapid and alarming spread of Plasmodium falciparum (pf) drug resistance shed light on two main concerns: (i) The limited number of therapeutic targets addressed by current antimalarials, and subsequently; (ii) The lack of knowledge of their mechanism of action. The parasite survival depends on the digestion of 3/4 of the hemoglobin contained in infected erythrocytes for supplying the amino acids needed for the parasite growth [3-5]. Four parasitic proteases families well known namely aspartic proteases, cysteine proteases, aminopeptidases and metalloproteases, involved in this metabolic process, are expressed during the erythrocytic stage of the parasite life cycle and their inhibition has proven to be central to avoid parasite proliferation [3-8] Falcipain-2 (FP2) and Falcipain-3 (FP3) are key papain-family (C1) Clan CA trophozoite cysteine proteases localized in the Digestive Food Vacuole (DFV) that cleave host native or denatured human hemoglobin. FP2, the most-expressed and best-studied enzyme among falcipains, is a promising target for novel antimalarial drugs development [9]. Michael acceptor-based Peptidyl Vinyl Sulfones (VSP) is well recognized specific irreversible inhibitors of FP2 which act through a covalent and steric obstruction of the substrate access to the catalytic site [10]. The active site of FP2 is large and contains four pockets, S1, S'1, S2 and S3, each pocket accommodating one substituent, P1, P'1, P2 and P3 of peptide substrate. Thus, the scissile bond is localized between the residues P'1 and P1 of the peptide and a cut that minimizes the energy of complex (protein-substrate) is obtained when the remaining substituents, P2 and P3, fit together perfectly with the remaining pockets S2 and S3. Previous Structure-Activity Relationships (SAR) studies highlighted that FP2 and related enzymes, react preferentially with peptidyl ligands containing large hydrophobic side chain at P2 position and, have a clear preference for substrate that contain a Leucine (Leu) residue at this position rather than Phenylalanine (Phe) or Valine (Val). In fact, the fragments in position P2 provides the basis for the specificity of the peptide inhibitors and the S2 subsite reflects the key determinant of selectivity of FP2 [11]. Logically, the most hydrophobic residue, namely Phe, should be more specific, but the observed effect is quite different. This preference would be due to the side chain of amino acid Ile 85 in the crystal structure of FP2 (PDB code: 3BPF monomer B) that creates a small protrusion in the S2 pocket preventing thus good fixation of the aromatic rings [12]. Conversely, a peptide ligand, containing a Leu residue in P2, slots comfortably into the S2 subsite for maximizing hydrophobic interactions while reducing energy costs due to unfavorable conformational changes of Ile 85. Changes at position P3 have relatively low impact on vinyl sulfone's activity [11] making this position suitable for the design of hybrid molecules based on artemisinin and endoperoxide [13-15]. Considering that VSP are potent specific inhibitors of FP2 and the Artemisinin Molecule (ART) and its derivatives possess exceptional pharmacokinetic profile, a series of hybrid inhibitors Artemisinin-Vinyl Sulfone (AVSH) obtained by linking ART and VSP through the 4-hydroxymethylbenzoic acid in P3 position have been synthesized and evaluated [13]. As indicated by the authors in their conclusions, there is space to improve the bi-functional molecules as the SAR for both activities is better understood. A way to get insight into the SAR of AVSH is to virtually evaluate the impact of ART derivatives in P3 position upon FP2 inhibition.
In this work, we start from the series of 6 Hybrid AVSH as training set to build one descriptor QSAR model of FP2 inhibition. As ART studies were carried out through Quantum Mechanics (QM) approach instead of Molecular Mechanics (MM) for Vinyl Sulfones (VS) against FP2, we decided to develop two FP2 - AVSH interaction QSAR models with one descriptor: the first one based on our MM-PB complexation and a second one based QM study of AVSH interaction with a short list of FP2 active site residues. The standard Gibbs Free Energy (GFE) change upon Enzyme:Inhibitor (E:I) complexes formation (ΔGcom) was computed and used to build a linear regression with experimental inhibition potency (IC50), in order to establish two quantitative structure-activity models (QSAR): MM-PB (pIC50, ΔGcom) and QM-PCM (pIC50, ΔGQMcom) QSAR. Each complex was built from a reference crystal structure of FP2 in complex with epoxysuccinate E64.
Methods
The complexation methodology has been described largely according to a procedure successfully used to elaborate one descriptor QSAR models of viral, bacterial and protozoal protease-inhibitor complexes and from them to design peptidomimetic, hydroxynaphthoic, thymidine, triclosan and pyrrolidine carboxamide derivative inhibitors [16-25].
Training and validation sets
A series of 6 Artemisinin-dipeptidyl Vinyl Sulfone Hybrid (AVSH) molecules used in this work were selected from the literature with activities measured in enzyme inhibition assays [13].
They cover a sufficiently broad range in order to allow a reliable QSAR model to be built.
Simulations were performed for enzyme: inhibitor (FP2:AVSH) complexes, free enzyme FP2 and free inhibitor AVSH from high-resolution crystal structure of the FP2 complexed with inhibitor epoxysuccinate E64 (Protein Data Bank entry code 3BPF at resolution 2.9Å [8]) using Insight II molecular modeling program. First, no crystallographic water molecule was conserved into the molecular model then, the structures of FP2 and FP2:AVSH complexes were at pH 7 with neutral N- and C-terminal and finally, the protonizable and ionizable residues of FP2 were charged.
The inhibitors were constructed from E64 within the active-site of the reference crystal structure 3BPF by replacing the appropriate fragments or function groups (in situ modifications). All rotatable bonds of the replacing fragments were subjected to an exhaustive conformational search coupled with careful gradual energy-minimization of the modified inhibitor and active-site residues of FP2 located in the immediate vicinity (around 5Å radius) in order to identify low-energy bounded conformations of the modified inhibitor. The resulting low-energy structures of the E:I complexes were then carefully refined by energy-minimization procedure of the entire complex to obtain the stable structure of binary FP2:AVSH complexes.
Calculation of binding affinity
The complete description of the complexation binding affinity (ΔGcom) is described in ref. [25].
Describes the relative enthalpic contribution to the GFE change related to the intermolecular interactions in E:I complex derived by MM; and represent, respectively, the relative solvation GFE and simplified relative vibrational entropy.
Molecular mechanics
Molecular models of inhibitors, falcipain 2 enzyme and E:I complexes were carried out by Molecular Mechanics (MM) in all-atom representation using atomic, bond and charge parameters of class II consistent force field CFF91 [26]. In all molecular simulations performed by MM calculations, a relative permittivity of 4 was employed in order to take into account the dielectric shielding effect in proteins. Geometry optimizations and energy minimizations of E:I complexes, free E and I obtained by relaxing the molecular structures gradually, have been fully described in [25].
Conformational search
Conformations of free I was derived from its bound ones in the previously minimized binary complexes E:I by gradual relaxation to the nearest local minimum as described in [25].
Solvation Gibbs free energy
The Solvation Gibbs free energy treatment in this work has been fully described in [25].
Interaction energy
The Molecular Mechanic interaction energy (Eint) calculation protocol available in Discovery Studio 2.5 [27] was used to compute the non-bonded interactions (van der Waals and electrostatic interatomic potential terms) between two sets of atoms in the E:I complexes. All pairs of interactions (between each enzyme residue and inhibitor) of the total enzyme-inhibitor interaction energy were evaluated using CFF force field parameters with a relative permittivity of 4 [27]. In particular, the breakdown of Eint into active-site residues contributions shows in the detail the strength and level of the main individual interactions in order to allow a quantitative analysis, which permits identification of residues with highest contribution to the ligand binding, leads to identify structural modifications for favorable binding affinity and suggests molecular moieties in the inhibitor which carries the key features responsible for the biological activity [22].
3D-QSAR pharmacophore generation
The 3D-QSAR Pharmacophore (PH4) implemented in Discovery Studio 2.5 [27] has been used to generate all the PH4. The methodology has been fully described in [25].
ADME-related properties
The ADME-related properties of AVSH derivatives were calculated using QikProp software program of Schrödinger suite [28]. The treatment in this work has been fully described in [25].
Quantum calculations
The ab initio computations were performed according to standard molecular orbital calculation scheme in the same way on two quadri-processor Workstations with Gaussian 03 at 6-31G level of theory. Hartree-Fock method was used to produce geometries and energies. Geometry of the ligand and the catalytic and other key residues: Gln36, Cys42, His174 and Asn204 (an average of 89 heavy atoms) was fully optimized with respect to all degrees of freedom of the vinyl sulfone moiety while the artemisinin group was kept in an initially optimized conformation. Let's recall that these hybrid ASVH the artemisinin moiety is not involved in FP2 inhibition. Finally geometry optimization to a global minimum was further tested with frequency calculation. All the calculations in the solvent were performed using the Polarized Continuum Model (PCM) [29,30]. The unique QM calculation performed on FP2 was a study by Molecular Dynamic (MD) simulations using hybrid AM1d/MM and M06-2X/MM potentials to understand FP2 inhibition by E64 [31]. The cost of ab initio computations justifies that we focus only relevant active site residues and compare inhibitor bound conformations from both MM and QM approaches.
The standard GFE change upon the E:I complex formation in liquid phase computed by the Quantum Mechanics - Polarizable Continuum Model approach (QM-PCM) can be approximated again by following equation
leading to
is enthalpic component and stands for the QM solvation component based on charge distribution.
The HF/6-31G optimized geometries were then submitted to PCM single-point calculations with dielectric constant outside the cavity of 78.4 for water. The free energy of a solute is defined as the sum of four terms namely cavity formation energy (Gcav), electrostatic energy (Gel), dispersion energy (Gdis) and repulsion energy (Grep) [32].
Results and Discussions
The chemical structures and experimental inhibitory activities of homogenous set of 6 AVSH inhibitors against FP2 of pf, determined in the same laboratory, by Capela, et al. [13] are given in Table 1. Their observed activities cover a wide activity range from 0.35 to 22.4 µM, which is sufficient for building reliable QSAR models of FP2 inhibition.
One descriptor QSAR models
The relative complexation Gibbs free energy of E:I complex formation from free enzyme E and free inhibitor I (ΔΔGcom, see the methods section for definition), was computed for 3D molecular models prepared from high resolution reference crystal structure of FP2:E-64 complex (PDB entry code 3BPF [8]) by in situ modifications of the template inhibitor E-64 within the binding site of the enzyme, as described in the training and validation set section of this paper. Table 2 lists the computed values of Molecular Mechanics relative Gibbs free energies upon complex formation (ΔΔGcom) and its components (equation 1) while Table 3 gives the Quantum Mechanics calculated values using (equation 3) as described in the methods section (Quantum Mechanics). Since ΔΔGcom quantities were evaluated in approximate way, the relevance of the binding models was assessed using linear correlations between experimental activity data (half maximal inhibitory concentration and computed quantities according the following correlation equation:
Where a and b are regression coefficients.
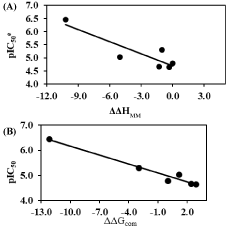
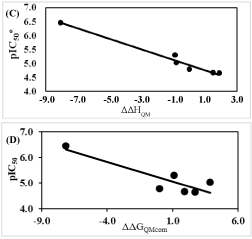
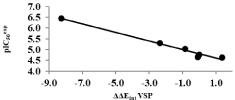
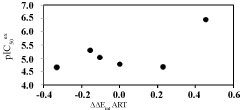
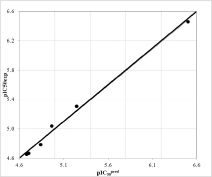
The regression analysis and the relevance statistical data of QSAR models, using classical and quantum descriptors, are given in Table 4 and Table 5 respectively and their corresponding correlation plots are displayed in Figure 1 and Figure 2. Two correlation equations obtained for GFE upon E:I complex formation ΔΔGcom and its enthalpic component ΔΔHMM were assessed for each complexation QSARMM resp. QSARQM model; this last one for which the complex was limited to the ligand surrounded by the four closest active site residues (Gln36, Cys42, His174 and Asn204).
The QSARMM statistical data, in Table 4, documents the robustness of the model in the gas phase (ΔΔHMM) and finally in the aqueous medium (ΔΔGcom): R2 correlation coefficients 0.79 ≤ R2 ≤ 0.98; Leave-One-Out (LOO) cross validated R2 0.74 ≤ R2XV ≤ 0.93 and the Fischer test coefficient F = 14.97 respectively F = 112.52.
In the same way QSARQM statistical data from QM computations are listed in Table 5: R2 =0.97, R2XV = 0.96 and F = 126.45 for gas (ΔΔHQM). In the liquid phase ΔΔGQMcom; is dependent on ΔΔGQM_solv = ΔΔGcav+ ΔΔGel + ΔΔGdisp + ΔΔGrep: taking account of all the solvation terms resulted in a lack of correlation; the removal of electrostatic term (keeping cavitation, dispersion and repulsion) led to a linear correlation R2 = 0.77, R2XV = 0.72 and F = 13.71.
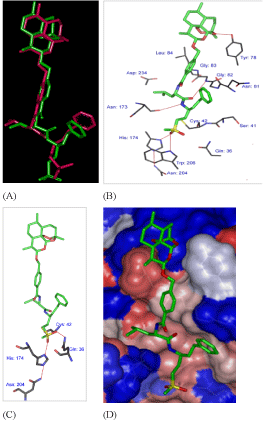
The bound conformation of the best active AVSH6 from QSARMM resp. QSARQM are superimposed in Figure 3A displaying the lowest RMSD value 1.51Å (see Table 6) of the range 1.51-3.35Å.
Binding mode of AVSH inhibitors
The binding mode of AVSHs at the active site of FP2, such as Hydrogen Bonds (HB), hydrophobic contacts upon pockets filling and Van der Waals interactions, all involving key FP2 residues, are conserved. Due to the peptidic structure of AVSHs with P'1, P1, P2 and P3, interactions with respective pockets will give structural information on binding affinity.
According to the AVSH's activity, Structure Activity Relationship (SAR) data classify the sequences in the order Leu-hPhe > Phe-hPhe > Phe-Phe and the P'1 occupancy in the order Ph > Me confirming the fact that the effect of substituents at the P'1 position seems to be dependent on the dipeptide core [13].
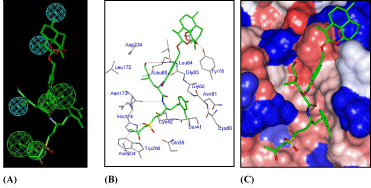
The binding mode of AVSH6 is depicted in Figure 3. In Figure 3B the detailed interactions with each pocket residue is displayed and hydrogen bonds in red line: P'1 (Asn 204 - His 174 and Trp 206), Asn 173, Gly 82 and the HB contact between Tyr 78 and Artemisinin moiety. The hydrophobic contacts in P'1, P1, P2 and P3 are in Table 7 where interaction energy breakdown to each active site residue contribution are listed.
In Figure 3C the interactions between AVSH6 and the selected active site residues computed through QM scheme are depicted such as HBs (Asn 204 - His 174, Gln 36). The correlation between the QM enthalpic contribution ΔΔHQM to GFE documents the accuracy of HF 6-31G and let look forward the benefit of its extension to other active residues. One fundamental question arises about the use of QM ab initio approach for the whole active site excluding the remaining of the enzyme usually subjected to MM in the so called QM/MM scheme.
Another subsequent arising deep concern is the QM solvation component of the GFE (ΔΔGQMcom) upon FP2-AVSHx complex formation: ΔΔGQM_solv. Indeed ΔΔGQM_solv = ΔΔGcav + ΔΔGel + ΔΔGdisp + ΔΔGrep according to an explicit-solvent approach oppositely to the implicit-solvent one we are used to with MM. QM/MM scheme accordingly considers the QM moiety as a solute in the MM surrounding solvent. The electrostatic contribution from Miertus, et al. [29,30] based on the Polarizable Continuum Method (PCM), is one of the most efficient evaluation of ΔΔGel since it takes into account a realistic molecular cavity shape. It is nevertheless tributary of the accuracy of the representation of continuous charge distribution over the solvent cavity surface as a set of single point charges [33]. Obviously a better evaluation of the point charges and of the subsequent electrostatic potential they produce will lead to better ΔΔGel and its inclusion in ΔΔGQM_solv.
The picture of AVSH6 in Figure 3D in the Connolly surface around AVSH6 showing the pockets confirms the preeminence of the sequence Leu-hPhe.
Interaction energy
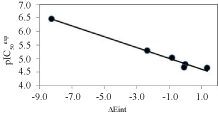
To identify the structural features of AVSHs and FP2 active site for an increase of FP2 inhibition, the computed MM enzyme - ligand interaction energy (Eint) as described above in methods section is a relevant descriptor. Various exploitation of Eint helped in the design of new potent analogs especially the Eint breakdown diagram to each active site residue individual contribution. We successfully, from its comparative analysis for most potent at one side and less potent training set molecules at the other, identified new R-groups and designed novel analogs with increased predicted activity [22]. The AVSH regression data of the global Eint vs. are listed in Table 8 and Table 9 and plotted in Figure 4.
AVSHs are hybrid molecules combining two pharmacophores (PH4) in a single ligand for a synergistic biological action on FP2. In order to get insight into each moiety contribution to FP2 - AVSHx interaction energy (Eint), the correlation of Eint_ART resp. Eint_VSP versus biological activity are listed in Table 10 and displayed in Figure 5 resp. Figure 6. As expected VSP moiety carries the whole inhibition power [13]. Despite ART moiety is not directly involved in the Eint correlation vs. , ART moiety's substitutions may partially impact the FP2 - AVSHx affinity since Eint is estimated in gas phase and at the enthalpic level only. The GFE including the entropy loss and the solvent effect upon complex formation in the QSAR model will be a better descriptor to evaluate the ART substituted analogs [34].
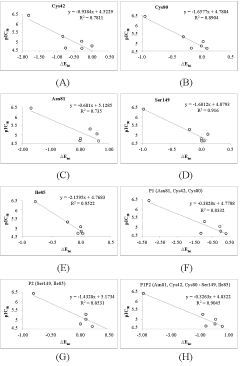
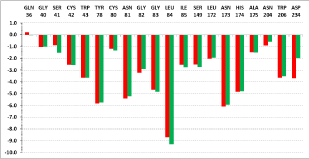
The cartography of Eint from each active site residues contribution (in % of Eint) is presented in Table 7 and classified by pockets. S1 and S2 pockets contributions are in trend of biological activity while S'1 and S3 are in the opposite trend suggesting that the best substitutions for novel analogs are expected in P1 and P2. To confirm this, a deeper analysis of the cartography is displayed in Figure 7 and Figure 8. Key S1 residues Cys42 (Figure 7A), Cys80 (Figure 7B) and Asn81 (Figure 7C) contribution correlate with individually and collectively (Figure 7F). In the same way S2 ones Ile85 (Figure 7D) and Ser149 (Figure 7E) individually and cumulatively (Figure 7G) do. S1 and S2 key residues contribution correlates slightly more with (Figure 7H).
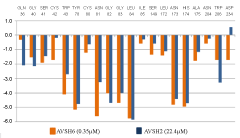
Finally Eint breakdown comparative analysis diagram for the best active AVSH6 and the less active AVSH2 is displayed in Figure 8. The prominent role of S1 (Cys42, Cys80, Asn81) and S2 (Ile85, Ser149) above cited key residues clearly is justified. Moreover contributions from Gln36, Gly40 and Trp206 may be improved for greater affinity.
From the reliable QSAR model using Eint parameters, the computed breakdown of Eint into contribution of each FP2 active site residue for AVSH2 (22.4 µM) and AVSH6 (0.35 µM) is depicted in Figure 8. The comparative analysis limited to these active site residues clearly justifies the potency divide between the best TS AVSH6 and the lowest AVSH2 particularly for Cys42, Asn81, Ser149 and Asp234.
3D-QSAR pharmacophore model of FP2 inhibition
Pharmacophore (PH4) modeling assumes that a set of structural features in a ligand is recognized by the receptor active site as "a hand in glove" and is responsible for biological activity. Bound conformations of AVSH inhibitors taken from the model of complexation were used for computing of the 3D-QSAR pharmacophore models of inhibitory activity by means of Catalyst Hypogen algorithm implemented in Discovery Studio 2.5. Top scoring or best pharmacophore hypothesis was obtained in three phases, as we described in methods section, from a set of most active AVSHs inhibitors.
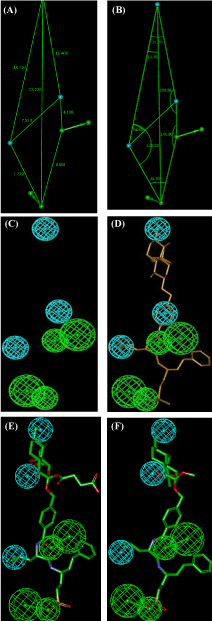
The statistical indicators such as the Root-Mean-Square Deviation (RMSD), squared correlation coefficient (R2) and costs of the ten best hypotheses generated are listed in Table 11 and the geometry of best of the best hypotheses (Hypo1) of AVSHs inhibition is displayed on Figure 9. To check the consistency of created pharmacophore model, the regression equation for vs. from Hypo1:
is also plotted in Figure 10.
New FP2 AVSH inhibitors analogs of FP2
Analogs from ART moiety substitution
The design of novel AVSH analogs is based on the following strategy: "Does the artemisinin moiety perform better than its derivatives"? In order to bring relevant answer we have virtually evaluated ART known derivatives: Dihydro-artemisinin, DihydroArtemether, DihydroArteether and DihydroArtesunate. Twenty four (24) novel analogs carrying the same VSP moiety of AVSH have been derived according to this scheme; they are listed in Table 12. The virtually computed activity , the GFE and its components for the 24 analogs are listed in Table 13. The AVSH6 analogs (6F-Dihydro, 0.17 µM), (6F-Arteether, 0.05 µM) and (6F-Artemether, 0.07 µM) are respectively twice, seven and five times more potent than the best training set AVSH6 (0.35 µM).
Analogs from VSP moiety substitutions
The main idea in the design of novel AVSH analogs is the substitutions at R1, R2 and R3 positions of the VSP moiety. Since the QSAR training set fully populated by AVSHx substituted at these three subsites, the R-groups suggested in Table 14 are intended to fill the pockets and improve the affinity with FP2 according to the differential analysis of Eint breakdown to each active site residue contribution (Eint_ASR). Figure 11 displays the Eint_ASR for the best designed analogs (1-5-4, 0.5 nM) and (1-9-4, 0.8 nM) for the same residues as for Figure 8 where the best active TS AVSH6 and the less active AVSH2 are compared.
Residues for which the affinity (Eint_ASR) with AVSH2 is greater than with AVSH6
Gln36, Gly40, Gly82 and Trp206 in Figure 8: For these residues, the less active TS AVSH2 is in higher affinity than the best active AVSH6. The substitutions at R1, R2 and R3 position (Table 14) intended to improve the affinity resulted in additional and higher contributions as displayed in Figure 11 for the best analogs 1-5-4 and 1-9-4 (Table 15).
Other active site residues
The affinity is greater than each Eint_ASR value computed for the best TS active AVSH6.
The best designed analog 1-5-4 (0.5 nM) is a modified Leu-hPhe core of AVSH6: R1 (CH2CH2Ph) → ((CH2)2-Ph-oCl-pCH3), R2 (CH2CH(CH3)2) → (CH2CH(Me)(CH2)2OH) and R3 (CH3) → (CH2-COCH3) aiming at better fill the pockets. 1-5-4 mapping to the FP2 inhibition PH4 derived from QSARMM is depicted in Figure 12 indicating that all the features are occupied as in the case of AVSH6 (Figure 9D). In the same Figure 12 displaying the close up of 1-5-4 at FP2 active site, all HB formerly noticed for ASVH6 (Figure 3B) are conserved with higher strength. In the same way 1-9-4 (0.8 nM) where R1 (CH2CH2Ph) → ((CH2)2-Ph-oCl-pCH3), R2 (CH2CH(CH3)2) → (CH2CH(Me)(CH2)2OCl) and R3 (CH3) → (CH2-COCH3) is a slight modification from 1-5-4 since the hydrogen atom of the hydroxyl group of R2 is replaced by a chlorine (Figure 11 and Table 12).
ADME profile of new AVSH analogs
WHO expectations about forthcoming antimalarials include their low cost and their oral bioavailability. Table 16 displays the computed ADME profile of the vinyl moiety of predicted best active AVSH analogs. Since the AVSH are hybrid compounds the ADME profile of the global molecule displays high number of stars indicating that many of the descriptors values fall outside the range for 95% known drugs. The profile of artemisinin is a zero star one confirming that the ART moiety clearly has a positive profile with almost one hundred percent HOA (95.8). The profile of the second moiety, the vinyl one, comes from Table 16: Their %HOA(r) are low for some of them for others, they are in the range for 95% of drugs (< 25% - poor, > 80% high) indicating middle range oral bioavailability. Predicted apparent Caco-2 cell membrane permeability in Boehringer- Ingelheim scale, the range for 95% of drugs being (< 25 poor, > 500 great) (n) in Table 16 confirms their poor membrane permeability but this has to be conjugated with the high permeability of Artemisinin (1886). The ADME profile for the best designed new analogues displays %HOA in a higher range for their majority with high cell membrane permeability. Novel designed AVSH drug likeness defined as the number of stars standing for the number of property descriptors (from 24 out of the full list of 49 descriptors of QikProp, ver. 3.7, release [28]) that fall outside of the range of values for 95% of known drugs, is low and in the same range as that for some current antimalarials of the Artemisinin Combined Therapy (ACT) initiative. The ADME profile of these hybrid antimalarials has to be analyzed as for ACT moieties i.e. two separated molecules bearing complementary profiles.
Conclusion
The FP2 inhibition pharmacophore combining Artemisinin and vinyl sulfone known features has served in the design of novel AVSH analogs with favorable pharmacokinetic profile by taking benefit from structural information revealed by complexation QSARMM model. Despite the Artemisinin moiety's mechanism of action does not target the protease, the global designed hybrid AVSH better fills the active site pockets, producing stabilizing interactions, exemplified in the best designed analogs (1-5-4, 500 pM) and (1-9-4, 800 pM) displaying predicted IC50 hundreds times higher than that of the best training set AVSH6. Although these predicted values look too optimistic, they are worth being proposed for synthesis and evaluation. In the same way an attempt of QSARQM 6-31 G model elaboration with active site key residues only resulted in a similar binding mode of AVSH at FP2 active site with a predicted IC50 of 170 nM (from the enthalpic component, Table 5, eq. (C)) for 1-5-4, twice more potent than AVSH6 and of 160 nM from the complexation (Table 5, eq. (D)). This QM result, despite the approximate way the approach was conducted, is the same trend as those from MM complexation model. Undoubtedly taking into account the whole FP2 active site and a better evaluation of the electrostatic (solvation) contribution to GFE will yield a better result and shed light on more accurate prediction.
Conflicts of Interest
The authors declare there is no 'Conflict of Interest' in relation with the work presented herein.
Acknowledgement
The authors thank the Computing Unit of the University Nangui Abrougoua (UNA) for computing facilities. This work was also supported by the Slovak Research and Development Agency grants APVV-14-0294 and APVV-14-0113".
References
- http://www.who.int/en/.
- Wongsrichanalai C, Meshnick SR (2008) Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border. Emerg Infect Dis 14: 716-719.
- Francis SE, Sullivan DJ Jr, Goldberg DE (1997) Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol 51: 97-123.
- Rosenthal PJ (2002) Hydrolysis of erythrocyte proteins by proteases of malaria parasites. Curr Opin Hematol 9: 140-145.
- Goldberg DE, Slater AF, Cerami A, et al. (1990) Hemoglobin degradation in the malaria parasite Plasmodium falciparum: an ordered process in a unique organelle. Proc Natl Acad Sci U S A 87: 2931-2935.
- Rosenthal PJ (2004) Cysteine proteases of malaria parasites. Int J Parasitol 34: 1489-1499.
- Hogg T, Nagarajan K, Herzberg S, et al. (2006) Structural and functional characterization of Falcipain-2, a hemoglobinase from the malarial parasite Plasmodium falciparum. J Biol Chem 281: 25425-25437.
- Kerr ID, Lee JH, Pandey KC, et al. (2009) Structures of falcipain-2 and falcipain-3 bound to small molecule inhibitors: implications for substrate specificity. J Med Chem 52: 852-857.
- Bekono BD, Ntie-Kang F, Owono Owono LC, et al. (2017) Targeting cysteine proteases from Plasmodium falciparum: A general overview, rational drug design and computational approaches for drug discovery. Curr Drug Targets 18.
- Santos MM, Moreira R (2007) Michael acceptors as cysteine protease inhibitors. Mini Rev Med Chem 7: 1040-1050.
- Shenai BR, Lee BJ, Alvarez-Hernandez A, et al. (2003) Structure-activity relationships for inhibition of cysteine protease activity and development of Plasmodium falciparum by peptidyl vinyl sulfones. Antimicrob Agents Chemother 47: 154-160.
- Wang SX, Pandey KC, Somoza JR, et al. (2006) Structural basis for unique mechanisms of folding and hemoglobin binding by a malarial protease. Proc Natl Acad Sci U S A 103: 11503-11508.
- Capela R, Oliveira R, Goncalves LM, et al. (2009) Artemisinin-dipeptidyl vinyl sulfone hybrid molecules: design, synthesis and preliminary SAR for antiplasmodial activity and falcipain-2 inhibition. Bioorg Med Chem Lett 19: 3229-3232.
- Kerr ID, Lee JH, Farady CJ, et al. (2009) Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J Biol Chem 284: 25697-25703.
- Oliveira R, Newton AS, Guedes RC, et al. (2013) An endoperoxide-based hybrid approach to deliver falcipain inhibitors inside malaria parasites. ChemMedChem 8: 1528-1536.
- Frecer V, Kabelac M, De Nardi P, et al. (2004) Structure-based design of inhibitors of NS3 serine protease of hepatitis C virus. J Mol Graph Model 22: 209-220.
- Frecer V, Jedinak A, Tossi A, et al. (2005) Structure Based Design of Inhibitors of Aspartic Protease of HIV-1. Letters in Drug Design & Discovery 2: 638-646.
- Frecer V, Berti F, Benedetti F, et al. (2008) Design of peptidomimetic inhibitors of aspartic protease of HIV-1 containing -Phe Psi Pro- core and displaying favourable ADME-related properties. J Mol Graph Mod 27: 376-387.
- Dali B, Keita M, Megnassan E, et al. (2012) Insight into selectivity of peptidomimetic inhibitors with modified statine core for plasmepsin II of Plasmodium falciparum over human cathepsin D. Chem Biol Drug Des 79: 411-430.
- Megnassan E, Keita M, Bieri C, et al. (2012) Design of novel dihydroxynaphthoic acid inhibitors of Plasmodium falciparum lactate dehydrogenase. Med Chem 8: 970-984.
- Owono Owono LC, Keita M, Megnassan E, et al. (2013) Design of Thymidine Analogues Targeting Thymidilate Kinase of Mycobacterium tuberculosis. Tuberc Res Treat 1-13.
- Keita M, Kumar A, Dali B, et al. (2014) Quantitative structure-activity relationships and design of thymine-like inhibitors of thymidine monophosphate kinase of Mycobacterium tuberculosis with favourable pharmacokinetic profiles. RSC Advances 4: 55853-55866.
- Frecer V, Seneci P, Miertus S (2011) Computer-assisted combinatorial design of bicyclic thymidine analogs as inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J Comput Aided Mol Des 25: 31-49.
- Owono Owono LC, Ntie-Kang F, Keita M, et al. (2015) Virtually Designed Triclosan-Based Inhibitors of Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis and of Plasmodium falciparum. Molecular Informatics 34: 292-307.
- Kouassi AF, Kone M, Keita M, et al. (2015) Computer-Aided Design of Orally Bioavailable Pyrrolidine Carboxamide Inhibitors of Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis with Favorable Pharmacokinetic Profiles. Int J Mol Sci 16: 29744-29771.
- Maple JR, Hwang MJ, Stockfisch TP, et al. (1994) Derivation of class II force fields. I. Methodology and quantum force field for the alkyl functional group and alkane molecules. Journal of Computational Chemistry 15: 162-182.
- Discovery Studio molecular modeling and simulation program (2009) version 2.5. Accelrys, San Diego, California, USA.
- QikProp (2014) Version 3.7, Release 14. X Schrödinger, LLC. New York, USA.
- Miertuš S, Scrocco E, Tomasi J (1981) Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chemical Physics 55: 117-129.
- Miertuš S, Tomasi J (1982) Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chemical Physics 65: 239-245.
- Arafet K, Ferrer S, Marti S, et al. (2014) Quantum mechanics/molecular mechanics studies of the mechanism of falcipain-2 inhibition by the epoxysuccinate E64. Biochemistry 53: 3336-3346.
- Frisch MJ, Trucks GW, Schlegel HB, et al. (2003) Gaussian 03, Revision B.05. Pittsburgh, Pennsylvania.
- Leach RA (2001) Molecular Modelling, Principles and Applications. (2nd edn), chapter 11, Pearson Education.
- Freire E (2008) Do enthalpy and entropy distinguish first in class from best in class?. Drug Discov Today 13: 869-874.
Corresponding Author
Aubin N'Guessan, Laboratoire de Physique Fondamentale et Appliquée, UFR SFA, University Nangui Abrogoua, 02 BP 801, Abidjan 02, Cote D'Ivoire, Tel: +225-02-36-30-08.
Copyright
© 2017 N'Guessan A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.