The Genetic Ablation of TNF-α Attenuates Wnt-Signaling and Adiposity in High Fat Diet-Induced Obese Mice
Abstract
A chronic low-grade inflammation is considered as a consequence of obesity, and linked with multiple complications. However, it is under-investigated how inflammatory cytokines mediate adipogenesis. This study investigated the role of Tumor Necrosis Factor α (TNF-α) on adipogenes is over high-fat diet feeding. Three groups of wild type or TNF-α-/-mice with the same C57BL/6 genetic background were utilized in this study: wild type fed with a low-fat diet (WT-LFD), wild type fed with a high-fat diet (WT-HFD), and TNF-α-/- fed with a HFD (TNF-HFD). After 16-wk feeding, inflammatory cytokine, Wnt pathway and adipogenesis-related genes were analyzed. HFD feeding increased body weight in both WT-HFD and TNF-HFD groups, but genetic ablation of TNF-α attenuated HFD-induced obesity. In visceral adipose tissues, HFD elevated Wnt/β-catenin signaling, indicated by decreased phospho-GSK3β and active β-catenin, two key components within the Wnt pathway, and dysregulated adipogenesis, indicated by reduced PPARγ/CEBPα expressions. Whereas, the deletion of TNF-α suppressed Wnt-signaling, and restored expressions of adipogenes is-related genes, which were otherwise decreased in the HFD-induced obese animals. These findings demonstrated a critical role of TNF-α in the regulation of Wnt-signaling and adiposity in mice over a HFD feeding, indicating HFD-induced adipocyte dysfunction could be mitigated by targeting TNF-α and Wnt-signaling.
Keywords
Obesity, Inflammation, TNF-α, Wnt pathway, Adipogenesis, PPARγ
Introduction
It is well-established that obesity, with inflammation as one of the critical mechanisms, linked to a range of chronic diseases such as type 2 diabetes mellitus (T2DM), nonalcoholic fatty liver disease (NAFLD), cardiovascular disease (CVD), and cancer [1]. Obesity is well-accepted as a chronic inflammatory condition as a consequence of hypertrophic adipocyte expansion [2]. Whereas, how inflammatory cytokines reversely mediate adipogenesis is less investigated. To understand this mechanism and prevent chronic over nutrition-induced adipocyte dysfunction represents a promising approach to diminish the epidemic of obesity and its associated medical complications in our society [3,4].
Adipose tissue, as an endocrine organ, plays an important role in maintaining the whole-body energy metabolism [5]. In order to maintain its function, a constant differentiation of adipocytes (adipogenesis) is necessary in adipose tissue [6]. During the adipogenesis from undifferentiated pre-adipocyte to lipid-loaded mature adipocytes, Wnt/β-catenin signaling participates in the transition and aberrant over-activation of Wnt-signaling results in the interruption of this process by inhibiting PPARγ expression [7,8]. Among many transcriptional factors, PPARγ is one of the major transcriptional factors to orchestrate the adipogenesis. PPARγ regulates lipid and glucose metabolism, and PPARγ target genes critically involve in lipid uptake and adipogenesis [9]. PPARγ agonists have been used in the treatment of hyperlipidemia and hyperglycemia [10], and it has been implicated in the pathology of numerous diseases including cardiovascular diseases and cancer [11]. Therefore, it is important to understand the regulation of PPARγ expression in obesity, thereby providing feasible avenues to properly manage adipogenesis and related complications.
Tumor necrosis factor-alpha (TNF-α), which is one of pro-inflammatory cytokines found to be increased in obesity, has been considered an important mediator that causes metabolic dysregulations in the body via adipose tissue dysfunction [12]. Previous animal studies with TNF-α knockout (TNF-α-/-) mice showed that TNF-α deficiency attenuated insulin resistance in peripheral tissues and prevented excessive hepatic lipid accumulation in diet-induced obese mice [13-16]. However, the cellular mechanism(s), by which TNF-α medicates adipose dysfunction has not been clearly defined. This study aimed to investigate the role of TNF-α in the regulation of Wnt/β-catenin signaling in adipogenesis in mice over HFD feeding. We hypothesized that obesity-promoted TNF-α activates Wnt/β-catenin signaling in adipose tissue, and that its constant activation inhibits the production of PPARγ, thereby leading to further adipocyte dysfunction.
Methods
Animals and experimental diets
TNF-α-/- mice were purchased from Jackson laboratory (Bar Harbor, ME, USA). It was originally created on 129S/6 background, but has been backcrossed to C57BL/6 for >10 generations. Mice were housed under conditions of controlled temperature (22 °C - 25 °C) and illumination (12:12 hour light-dark cycle, lights on between 7:00 and 19:00) with food and water provided ad libitum. The animal use protocol was approved by the Institutional Animal Care and Use Committee of the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University and the Institutional Animal Care and Use Committee of the University of Massachusetts, Amherst.
All the C57BL/6 wild type and TNF-α-/- mice were initially fed a standard rodent chow diet (Teklad#8604, Envigo, Madison, WI). At 8 weeks of age, animals were divided into 3 groups (26 animals (13 males and 13 females) /group): wild type fed with a low-fat diet (WT-LFD, 10 % of total calorie from fat), wild type fed with a high-fat diet (WT-HFD, 60 % of total calorie from fat), and TNF-α-/- fed with the same high-fat diet (TNF-HFD).A high fat diet, which is commonly used to induce obesity, was purchased from Research Diets (HFD: D12492 vs. LFD: D12450B, New Brunswick, NJ). The dietary compositions were shown in Table 1. As significant gender differences in body weight gain and metabolic parameters exist with male mice more sensitive to high fat induced-induced obesity [17,18], in addition to the body weight and body composition, only male animals were used for molecular analysis in this study to avoid the confounding from gender. At the end of the experiment, mice were euthanized by CO2 asphyxiation followed by cervical dislocation. Exsanguination was performed by cardiac puncture and blood was collected. The abdomen was opened and epididymis fats were collected into foil packets and frozen in liquid N2 for subsequent RNA and protein assays.
Inflammatory cytokine assays
The inflammatory cytokines were measured by a chemiluminescence assay using the QuickPlex SQ 120 (Meso Scale Diagnostics, Rockville, MD). Assays were performed according to the manufacturer's instructions. Briefly, the protein samples including inflammatory cytokines were isolated from visceral fat tissues with lysis buffer prepared according to the protocol for the electrochemiluminescence assay (Meso Scale Discovery, Rockville, MD). Then 25 μl of calibrator standards or samples were added to each well of a 96-well plate, on the bottom of which antibodies for 3 cytokines (IFN-γ, IL-1β, and IL-6) were coated. After washing for 3 times, 50 μl of the detection antibody solution was added to each well. A four-parameter logistic fit curve was generated for each analyte using the standards and the levels of inflammatory cytokines in samples were calculated accordingly. Cytokines are express as ng of cytokine per mg protein. All standards and samples were measured in duplicate.
Real-time PCR for gene expression
Total RNA was extracted with the Trizol reagent (Invitrogen, Carlsbad, CA). The concentration as well as purity of RNA samples was determined by NanoDrop 2000 (Thermo Scientific, Waltham, MA). The first-strand cDNAs were synthesized from 0.5 μg total RNAs using QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA). Real-time PCR was performed on the ViiA 7 Real-Time PCR System (Applied Biosystems, Carlsbad, CA) using the SYBR green PCR reagent kit (Invitrogen, Carlsbad, CA) with the following thermal cycling conditions: 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 60 seconds. The cycle threshold (Ct) values were defined as the fractional cycle number at which the fluorescence passes an arbitrarily set threshold. The copy number of each transcript was calculated with respect to the GAPDH copy number. Amplification of specific transcripts was confirmed by analyzing melting curve profiles at the end of each PCR. Primer sequences and relative expressions of each genes examined are listed in Table 2.
Western blot analyses
The protein sample was isolated from adipose tissue using RIPA lysis buffer (Thermo Scientific, Waltham, MA) with protease inhibitor cocktail (Santa Cruz Biotechnology, Dallas, TX). The total protein concentrations were determined using a commercially available Pierce™ BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). Western blotting was performed to determine the protein levels of phospho-GSK3β and active β-catenin as well as adipogenesis-related genes Pparγ and Cebpα. Briefly, 40 μg of protein from each treatment was separated by 10% SDS-PAGE and transferred onto a PVDF membrane. After the nonspecific binding was blocked with 10% instant nonfat dry milk, membranes were then probed with specific primary antibodies overnight at 4 °C, followed by incubation with a horseradish peroxidase-conjugated secondary antibody (Bio-Rad, Hercules, CA). Anti-PPARγ, CEBPα, Phospho-GSK3β (Ser9) and GAPDH antibody was purchased from Cell Signaling Technology (Boston, MA, USA). Anti-Active-β-Catenin was purchased from EMD Millipore Corporation (Billerica, MA, USA). The chemiluminescence detection was achieved using ECL Plus Substrate (Amersham Biosciences, Waltham, MA), and band intensities were analyzed using Image J Version 1.49 (https://imagej.nih.gov). The ratios of the density of the proteins were calculated after normalizing to corresponding loading controls.
Statistical analysis
Data are expressed as means ± SEM, and statistical analysis was performed using SAS (Version 9.4, SAS Institute, Cary, NC). One-way ANOVA was applied to test the differences among the 3 groups. The expression of each gene was normalized to the housekeeping gene Gapdh (DCt = Cttarget gene-CtGapdh) Statistical analyses were performed based on ΔCt and relative expression is reported as 2−ΔΔCt, where ΔΔCt = DCt-Experiment-DCt-Control.
Results
Body weight and body composition
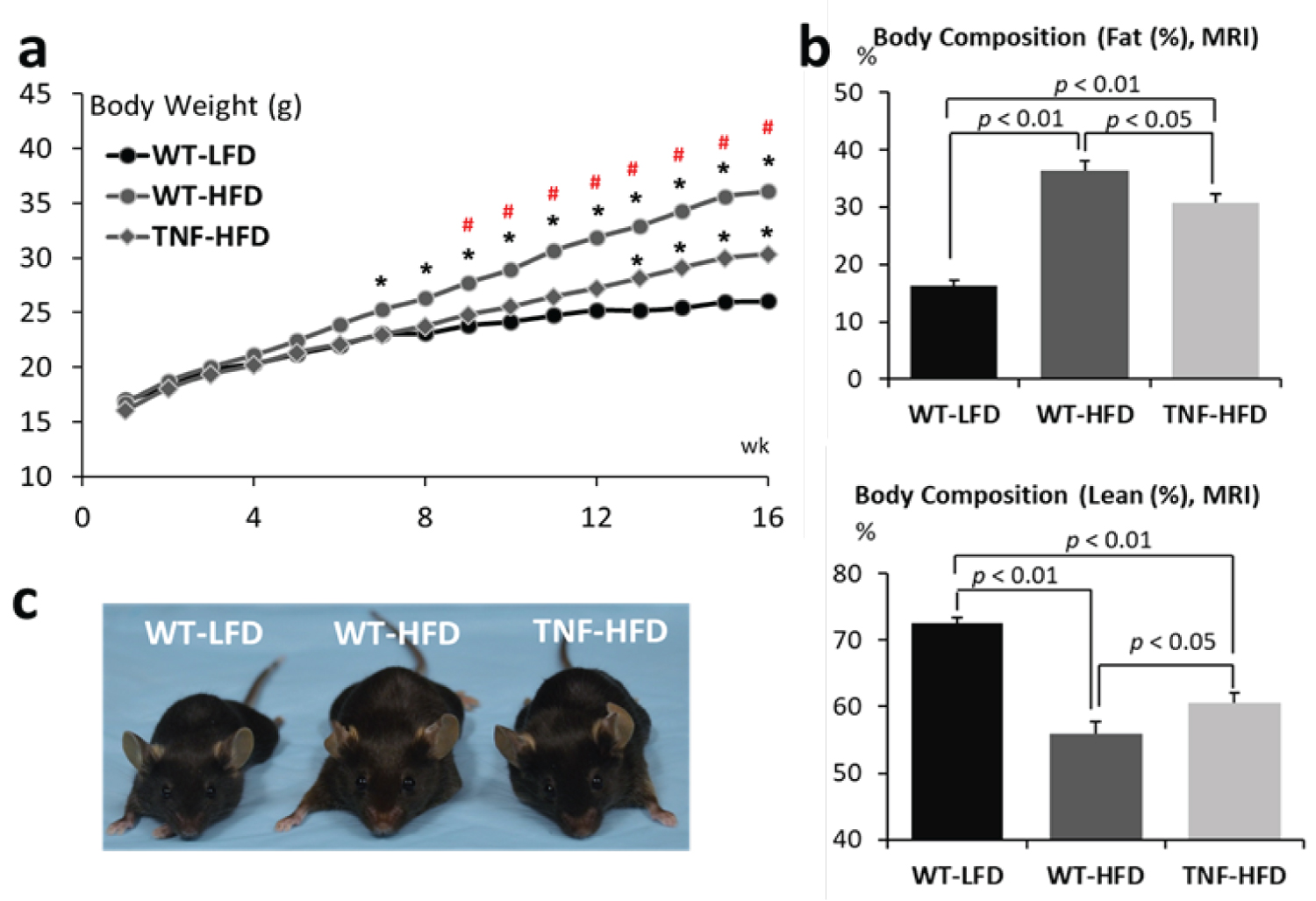
At the beginning of the experiment (8 wks of age), there were no differences across the 3 groups in terms of body weight. As expected, after 16 weeks of high fat diet feeding, the body weight of both wild type mice (WT-HFD) and TNF-α-/-(TNF-HFD) significantly increased when compared with that of wild type mice fed a low fat diet (WT-LFD). It is noteworthy that the genetic deletion of TNF-α reduced the sensitivity of high fat diet-induced obesity. Only after 7 weeks, the body weight of the wild type animals fed a high fat diet (WT-HFD) increased to a degree that was significantly higher than the body weight of those animals fed a low fat diet (WT-LFD), whereas a statistical difference was not observed for the TNF-α-/- animals (TNF-HFD) until 13 weeks of a high fat diet feeding (p < 0.05). The body weight of the WT-HFD group were significantly higher than that of the TNF-HFD group after feeding for the high fat diet for 9 weeks (p < 0.05). By the end of the experiment, the body weight gains were 53.7% for the WT-LFD group, 115.1% for the WT-HFD group, and 88.9% for the TNF-HFD group. Compared to the WT-LFD group, the body weights were 38.5% higher for the WT-HFD group, but only 15.5% for the TNF-HFD group (p < 0.05) ( Figure 1a).
The body composition data, measured by Magnetic Resonance Imaging (MRI), showed that, comparing to the WT-LFD group, the fat mass in the WT-HFD increased by 19.98% (16.35 ± 0.86% vs. 36.33 ± 1.82%, p < 0.01), but it only increased by 14.35% (30.70 ± 1.53% for the TNF-HFD, p < 0.01). It is also significantly different between the WT-HFD and TNF-HFD group (p < 0.05). On the contrast, the lean mass in the WT-HFD decreased by 16.48% (72.45 ± 0.78% vs. 55.97 ± 1.68%, p < 0.01), but it only decreased by 11.97% (60.48 ± 1.28% for the TNF-HFD, p < 0.01). It is also significantly different between the WT-HFD and TNF-HFD group (p < 0.05) (Figure 1b).
Inflammatory Cytokines
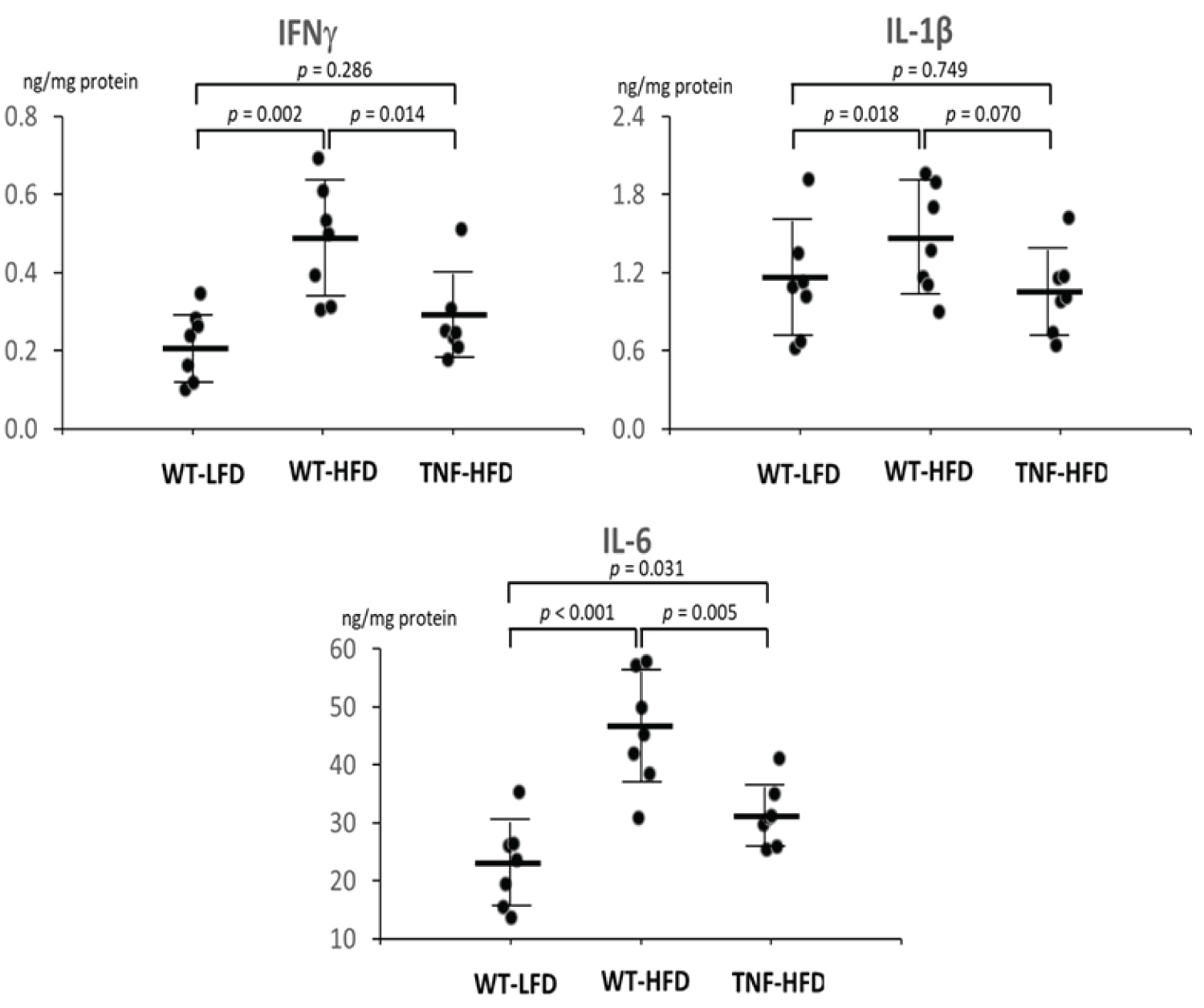
After observing the differentiated body weight change among groups, we measured 3 inflammatory cytokines in the visceral adipose tissue (IFNγ, IL-1β and IL-6). When compared to the WT-LFD group, the high fat diet (WT-HFD) significantly increased protein levels of IFNγ by 120.3% (p < 0.01), IL-1β by 29.5% (p < 0.05) and IL-6 by 100.6% (p < 0.001). However, the genetic ablation of TNF-α diminished the these elevations and the levels of IFNγ and IL-6 in the TNF-HFD group were significantly lower than in the WT-HFD groups (p < 0.05) with a numerical decrease of IL-1β (p = 0.07) (Figure 2).
The Wnt-signaling pathway
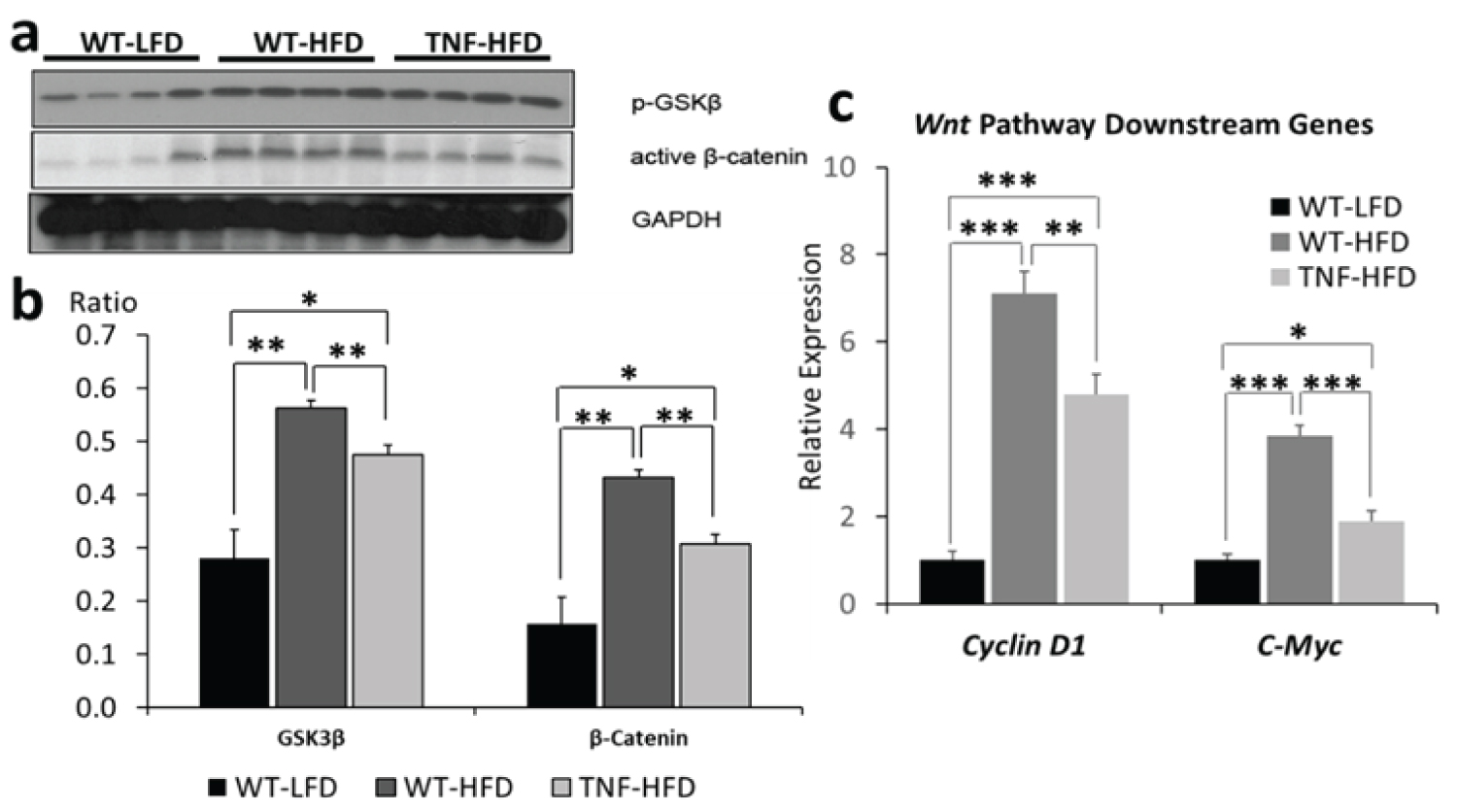
To investigate the Wnt-signaling pathway, we examined the protein levels of phosphorylated GSK3β and active β-catenin as well as the transcriptional levels of Wnt-signaling downstream target genes, C-Myc and Cyclin D1. As shown in ure 3a and ure 3b, when compared to WT-LFD, high fat diet (WT-HFD) significantly increased the protein levels for both phospho-GSK3β at the Ser 9 site (the inactive form) and active β-catenin (p < 0.01), whereas the genetic deletion of TNF-α (TNF-HFD) significantly diminished these effects (p < 0.01). After observing the mitigation of phosphorylated GSK3β and active β-catenin by TNF-α, we further examined the transcriptional expression of Wnt-signaling downstream genes. As shown in ure 3c,When compared to the WT-HFD group, the deletion of TNF-α (TNF-HFD group) reduced the expression of C-Myc and Cyclin D1 (p < 0.01) which were otherwise up regulated by high fat diet (p < 0.001, WT-LFD vs. WT-HFD).
Adipogenesis
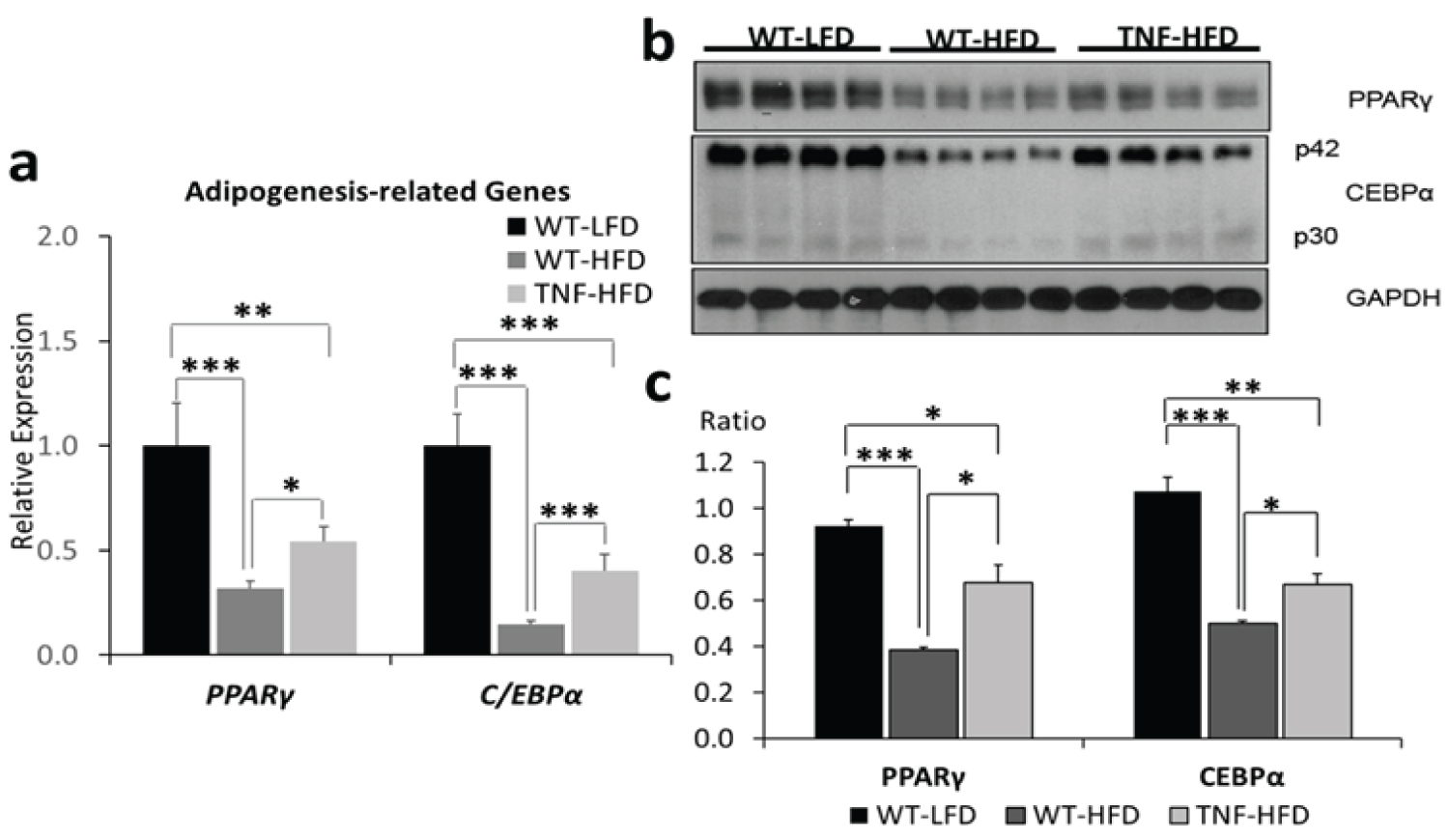
To investigate the influence of TNF-α on adipocyte dysfunction in high fat-induced obesity, we analyzed the expression, at both transcriptional and protein level, of PPARγ and CEBPα that are two critical transcriptional factors involved in adipogenesis. We found that, after a 16-wk high fat diet feeding, the transcriptional expression of Pparγ and Cebpα were significantly suppressed (p < 0.001) by high fat diet (WT-HFD vs WT-LFD), whereas the genetic deletion of TNF-α attenuated this effect (TNF-HFD vs WT-HFD, p < 0.05) (Figure 4). The analysis of the protein levels of these two adipogenesis–related genes recapitulated the transcriptional data (Figure 4).
Discussion
Obesity with adipocyte hypertrophic expansion represents a low-grade inflammatory condition [14,19-21], and the increased inflammatory cytokines are generally considered as a consequence of obesity. This study, on the contrast, demonstrated that genetic ablation of TNF-α attenuated HFD-induced obesity, potentially via attenuating Wnt/β-catenin signaling and restoring the adipogenic PPARγ/CEBPα expression, suggesting that TNF-α plays a causal role in adipocyte dysfunction during adipocyte hypertrophic expansion.
Since the initial discovery of escalated expression of TNF-α in adipose tissue by Hotamisligil and colleagues in 1993 [14], it is well-accepted that the adipose tissue is an active endocrine organ, which releases >50 different cytokines and adipokines [19,20]. In addition to the origins of inflammatory cytokine from adipocytes, adipocyte hypertrophy leads to the infiltration of immune cells, especially macrophages, and these immune cells significantly contribute to the production of pro-inflammatory cytokines [22]. Whereas, it is less understood how inflammatory cytokines influence adiposity, though a few pro-inflammatory cytokines have been shown possessing important metabolic effects, including effect on lipid metabolism [23,24].
Studies pertaining the role of TNF-α on adipocyte dysfunction and obesity are limited and results are highly inconsistent. The study performed by Uysal, et al. in 1997 [16] showed that the body weights were similar between the wild type and TNF-α-/- animals regardless feeding with a LF or HF diet throughout the 16-week study, but a significantly decreased body weight for the TNF-α-/- model fed a HF diet were observed in a study from others [13]. On the contrast, two other studies in 2012 [15] and 2016 [25] showed that TNF-α-/- mice were even prone to gain weight on HF diet when compared wild type animals. The study of Uysal, et al. [16] also showed that the genetic absence of TNF-α receptors did not attenuate body weight in genetically-induced obese animals (ob/ob) when compared to those ob/ob mice without the deletion of TNF-α receptors. However, another study [26] showed that the ablation of TNF-α receptor protected against diet-induced obesity. These inconsistent results require further investigation to understand the cellular action of TNF-α on adipogenesis.
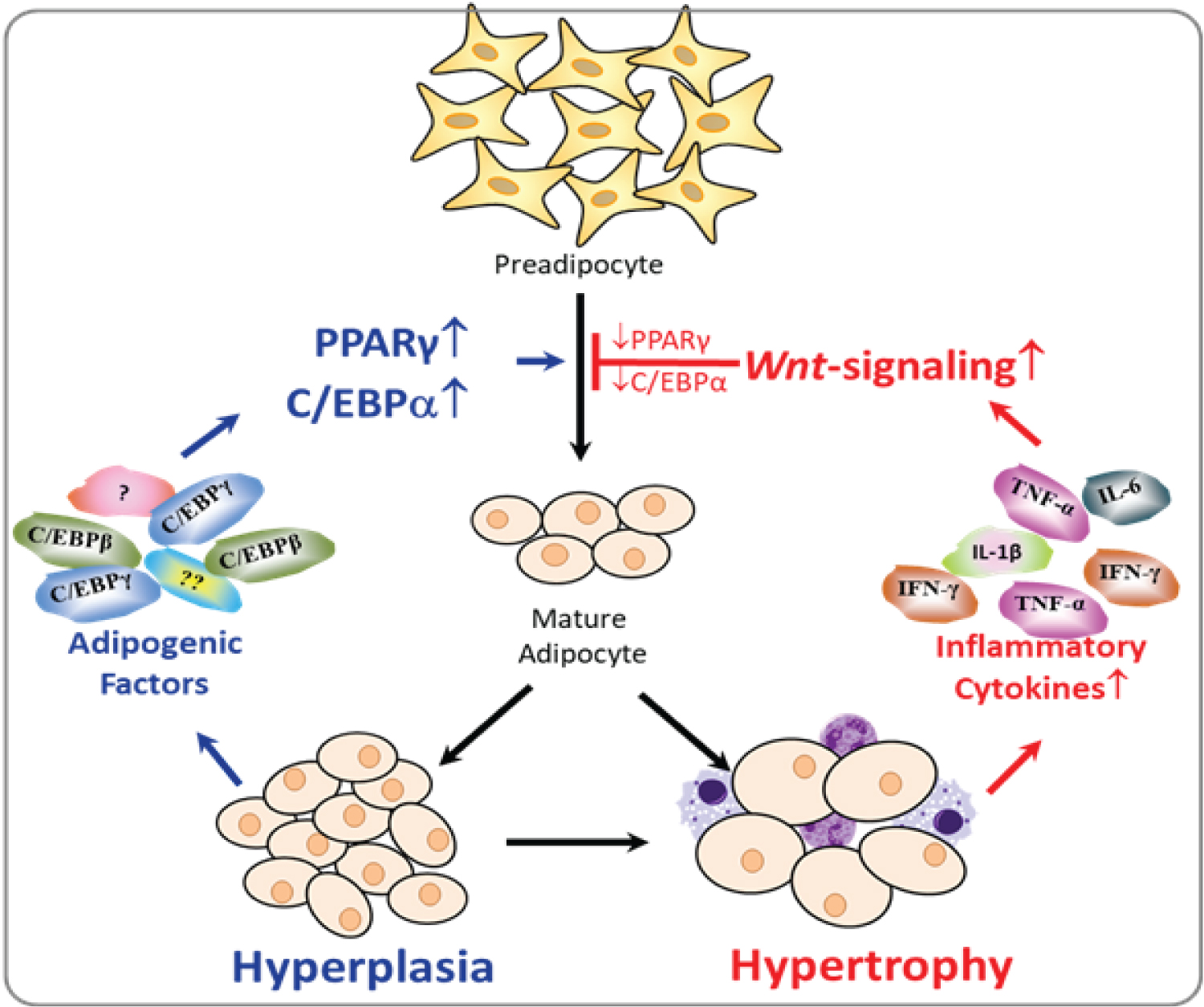
Diet-induced adiposity includes two stages: hyperplasia characterized by increased number of adipocytes, and hypertrophy featured with enlarged adipocytes. For a short-term over nutrition, our body may respond via stimulating the progression from pre-adipocyte to matured adipocyte, resulting in increased number of adipocytes called adipose hyperplasia. It is reported that, as early as 2 wks of HF diet feeding, an increased expression of PPARγ was observed in the liver tissue [27]. Whereas in chronic over nutrition, the over-uptake of lipids lead the adipocyte to the hypertrophic stage, which results in inflammation in the adipose tissue. Therefore, inflammatory cytokines may play regulatory roles on adipogenesis primarily in the latter hypertrophic stage when inflammation occurs (Figure 5). In our current study, animals were fed with a HFD (60 kcal% fat) for a long period (16 wks), and had reached the hypertrophic phase as indicated by increased inflammation. Our data clearly demonstrated that the genetic ablation of TNF-α attenuated Wnt/β-catenin signaling, restored the expression of PPARγ, and thereby suppressed HFD-induced obesity over a long-term HFD feeding. Our results may offer an explanation for the previous inconsistent results. TNF-α plays a regulatory role on adipogenesis primarily at the later hypertrophic stage when the inflammation occurs in the adipose tissue. In those previous studies, due to the either low fat content in the diet or the duration of feeding, animals in some of those studies might not reach the hypertrophic stage, and thereby resulted in inconsistent results.
Adipogenesis can be divided into two stages: Mesenchymal stem cells (MSCs) "commitment" to pre-adipocytes and pre-adipocytes differentiation into mature adipocytes. Initially, canonical Wnt-signaling controls the balance of MSC differentiating into myoblasts, osteoblasts or pre-adipocytes [28], and its activation suppresses commitment to the adipocytic lineage, probably suppressing the initiation of adiposity. This is manifested by the fact that genetic deletion of Wnt antagonist Sfrp1 leads to reduction of fat mass concomitant with increased bone mass [29]. On the contrast, our study demonstrated an impact of the canonical Wnt-signaling at the later stage of adipogenesis: The impairment of the proper progression from pre-adipocytes into matured adipocytes. Our results unraveled an important mechanism that TNF-α suppresses adipogenesis via enhancing the canonical Wnt-signaling, which consequently suppressed the adipogenic PPARγ/CEBPα expression, and promoted hypertrophic adipocyte expansion after 16-wk HFD feeding (Figure 5).
Conclusion
In summary, a chronic low-grade inflammation is generally considered as a consequence of obesity. On the contrast, our present findings demonstrated that the inflammatory cytokine TNF-α also causally influences adipose dysfunction. Once the adiposity reached the hypertrophic stage after 16-wk HFD feeding, the increased TNF-α activated Wnt/β-catenin signaling and interrupted adipogenesis as indicated by decreased expressions of adipogenic genes, PPARγ and CEBPα, and accompanying with the impaired adipogenesis is excessive adiposity and obesity. Whereas, the genetic ablation of TNF-α mitigate these alterations. These findings indicate that targeting TNF-α and Wnt/β-catenin signaling represents a feasible avenue to attenuate chronic obesity and its related medical complications.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
All authors declare no conflict interest.
Ethics Approval
The animal use protocol was approved by the Institutional Animal Care and Use Committee of the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University and the Institutional Animal Care and Use Committee of the University of Massachusetts, Amherst.
Funding Statement
This work was partially supported by the United States Department of Agriculture National Institute of Food and Agriculture (USDA/NIFA) grant (2014-67017-21762, ZL), USDA//NIFA Hatch project (1013548, ZL), Jean-Mayer Human Nutrition Research Center on Aging at Tufts University competitive intramural pilot grant (ZL).
References
- Weight H (2015) The health effects of overweight and obesity.
- Ellulu MS, Patimah I, Khaza'ai H, et al. (2017) Obesity and inflammation: The linking mechanism and the complications. Arch Med Sci 13: 851-863.
- Arner P, Bernard S, Salehpour M, et al. (2011) Dynamics of human adipose lipid turnover in health and metabolic disease. Nature 478: 110-113.
- Kloting N, Bluher M (2014) Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord 15: 277-287.
- Kershaw EE, Flier JS (2004) Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89: 2548-56.
- Spalding KL, Arner E, Westermark PO, et al. (2008) Dynamics of fat cell turnover in humans. Nature 453: 783-787.
- Lecarpentier Y, Claes V, Vallee A, et al. (2017) Interactions between PPAR Gamma and the Canonical Wnt/Beta-catenin pathway in type 2 diabetes and colon cancer. PPAR Res 2017: 5879090.
- Vallee A, Lecarpentier Y (2018) Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/beta-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front Immunol 9: 745.
- Jones JR, Barrick C, Kim KA, et al. (2005) Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A 102: 6207-6212.
- Lehrke M, Lazar MA (2005) The many faces of PPARgamma. Cell 123: 993-999.
- Kim JH, Song J, Park KW (2015) The multifaceted factor peroxisome proliferator-activated receptor gamma (PPARgamma) in metabolism, immunity, and cancer. Arch Pharm Res 38: 302-312.
- Guilherme A, Virbasius JV, Puri V, et al. (2008) Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9: 367-377.
- Bouter B, Geary N, Langhans W, et al. (2010) Diet-genotype interactions in the early development of obesity and insulin resistance in mice with a genetic deficiency in tumor necrosis factor-alpha. Metabolism 59: 1065-1073.
- Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 259: 87-91.
- Salles J, Tardif N, Landrier JF, et al. (2012) TNFalpha gene knockout differentially affects lipid deposition in liver and skeletal muscle of high-fat-diet mice. J Nutr Biochem 23: 1685-1693.
- Uysal KT, Wiesbrock SM, Marino MW, et al (1997) Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389: 610-614.
- Shi H, Clegg DJ (2009) Sex differences in the regulation of body weight. Physiol Behav 97: 199-204.
- Pettersson US, Walden TB, Carlsson PO, et al. (2012) Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS One 7: e46057.
- Fischer-Posovszky P, Wabitsch M, Hochberg Z (2007) Endocrinology of adipose tissue - an update. Horm Metab Res 39: 314-321.
- Tilg H, Moschen AR (2006) Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6: 772-783.
- Ramos EJ, Xu Y, Romanova I, et al. (2003) Is obesity an inflammatory disease? Surgery 134: 329-235.
- Weisberg SP, McCann D, Desai M, et al. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796-1808.
- Kunkel SL, Remick DG, Strieter RM, et al. (1989) Mechanisms that regulate the production and effects of tumor necrosis factor-alpha. Crit Rev Immunol 9: 93-117.
- Grunfeld C, Feingold KR (1991) The metabolic effects of tumor necrosis factor and other cytokines. Biotherapy 3: 143-158.
- Chen X, Gong Q, Wang CY, et al. (2016) High-fat diet induces distinct metabolic response in interleukin-6 and tumor necrosis factor-alpha knockout Mice. J Interferon Cytokine Res 36: 580-588.
- Romanatto T, Roman EA, Arruda AP, et al. (2009) Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J Biol Chem 284: 36213-36222.
- Inoue M, Ohtake T, Motomura W, et al. (2005) Increased expression of PPAR gamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun 336: 215-222.
- Christodoulides C, Lagathu C, Sethi JK, et al. (2009) Adipogenesis and WNT signalling. Trends Endocrinol Metab 20: 16-24.
- Bodine PV, Zhao W, Kharode YP, et al. (2004) The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol 18: 1222-1237.
Corresponding Author
Zhenhua Liu, Ph.D, Department of Nutrition, School of Public Health and Health Sciences, University of Massachusetts, Amherst, 100 Holdsworth Way, Amherst, MA, 01003, USA, Tel: 1-413-545-1075
Copyright
© 2020 Li J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.