TRANSCUP: A Scalable Workflow for Predicting Cancer of Unknown Primary Based on Next-Generation Transcriptome Profiling
Abstract
Cancer of unknown primary site (CUP) accounts for 5% of all cancer diagnoses. These patients may benefit from more precise treatment when primary cancer site was identified. Advances in high-throughput sequencing have enabled cost-effective sequencing the transcriptome for clinical application. Here, I present a free, scalable and extendable software for CUP predication called TRANSCUP, which enables (1) Raw data processing, (2) Read mapping, (3) Quality re-port, (4) Gene expression quantification, (5) Random forest machine learning model building for cancer type classification. TRANSCUP achieved high accuracy, sensitivity and specificity for tumor type classification based on external RNA-seq datasets. It has potential for broad clinical application for solving the CUP problem. TRANSCUP is open-source and freely available at https://github.com/plsysu/TRANSCUP
Keywords
CUP, Classification, Machine learning, RNA-seq, Workflow, Transcriptome
Introduction
The Cancer of unknown primary site (CUP) is a heterogeneous group of cancers in the absence of an identifiable primary tumor despite a standard diagnostic approach and accounts for approximately 5% of all cancer diagnoses [1]. The biologic mechanisms underlying this phenomenon are unknown [2]. CUP patients can be given site specific therapy with significant improvement in clinical outcome compared with empirical chemotherapy when cancer primary site was identified [3,4].
Currently, many molecular diagnostic methods have been widely applied in clinic, including RT-PCR [5], microRNA RT-PCR [6], gene expression microarray [7] and DNA methylation microarray [8]. Advances in high-throughput sequencing have enabled cost-effective sequencing the transcriptome for clinical application [9]. RNA-Seq based predication algorithm have been proposed using TCGA's RNA-Seq RSEM expression value and validated on both RNA-Seq and microarray dataset [10]. However, data analysis pipelines from raw FASTQ data to final tumor type predication are currently not fully implemented and validated computationally and are not publicly available [11].
Here, I present a free, scalable and extendable software for CUP predication called TRANSCUP, which comprises modules for raw data processing, read mapping, quality report, gene expression quantification and building a random forest model for cancer type classification. It achieved high accuracy, sensitivity and specificity for tumor type classification based on external RNA-seq datasets. It has potential for broad clinical application for solving the CUP problem. TRANSCUP is open-source and freely available at https://github.com/plsysu/TRANSCUP
Methods
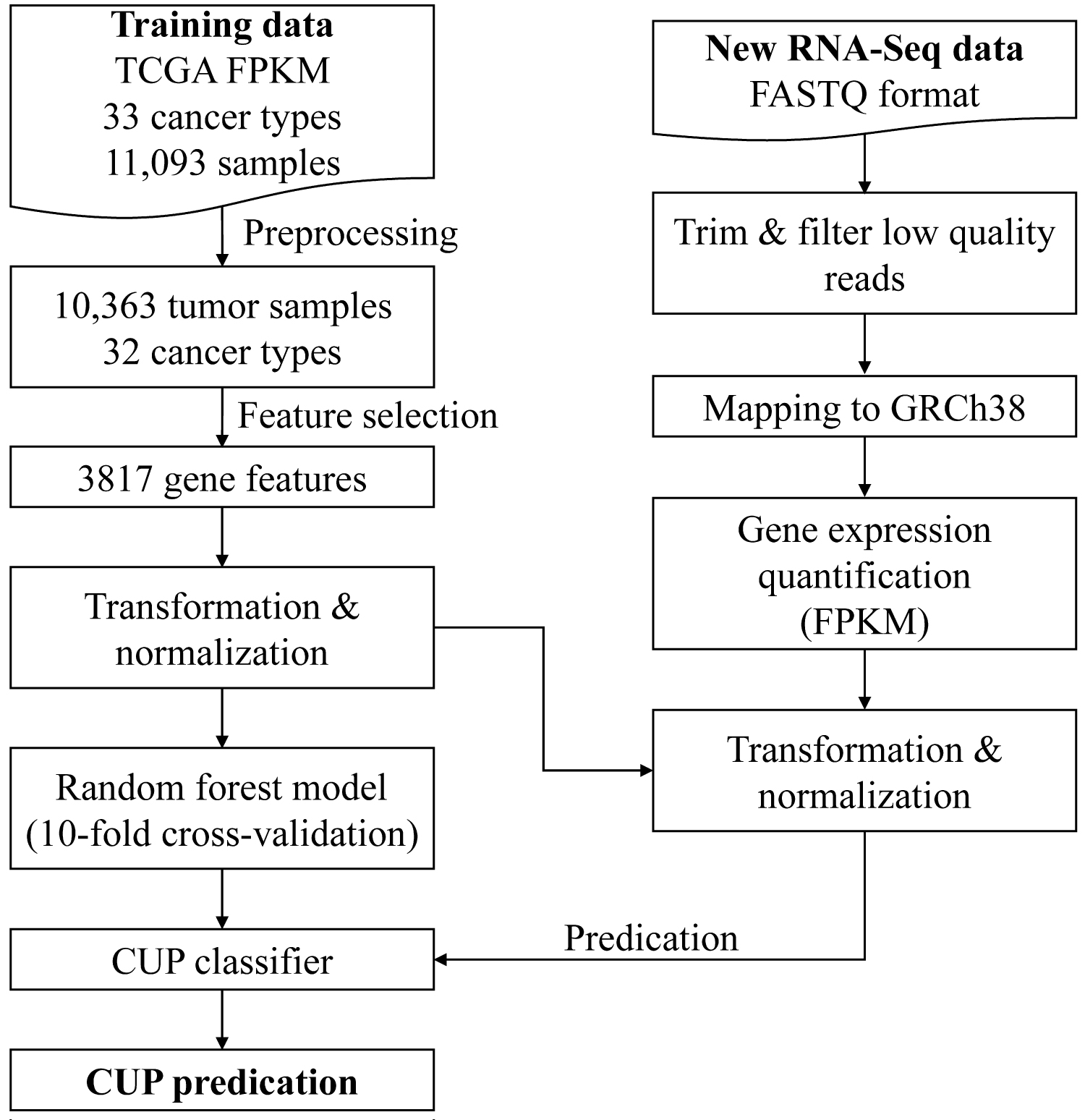
The TRANSCUP workflow is illustrated in Figure 1. To build CUP classifier, I use TCGA RNA-Seq data as training dataset, followed by sample and feature selection, data transformation, data normalization and random forest model building. To predict new tumor samples, I build a bioinformatics pipeline to process data from raw FASTQ reads to tumor type predication result. The details for implementation are the followings:
Training data source
TCGA harmonized gene expression quantification data and clinical data were retrieved from GDC portal (https://portal.gdc.cancer.gov/) via TCGAbiolinks [12]. Totally, 32 cancer types (COAD and READ were combined to CRC) and 10,363 tumor samples were used for model building.
Preprocess and feature selection
All FPKM expression data was log2-transformed, and only genes with (a) Maximum log2 expression greater than 2.5 and (b) Variance in log2 expression greater than 0.1 were retained. After filtering, the genes in each dataset were scaled to zero mean expression and unit variance. The mean and standard variance learned from training dataset for each gene was used to normalize the new data. Feature genes were selected by two criterion: 1) Those genes were different expressed by one tumor type to other tumor types; 2) Those genes were expressed more higher in one tumor type than other tumor types, which evaluated by t-test and log2-fold change, respectively. The top 100, 150, 200 genes' log2 fold change value or log2 fold change value larger than 2.5 or 3 were compared to find the best feature selection method.
Random forest model
The random forest algorithm was used for machine learning model building. To avoid overfitting, model was validated by 10-fold cross-validation. The final model was trained on all training data with the best hyper-parameters.
Bioinformatics pipeline
The raw reads were cleaned prior to following analysis by Trimmomatic [13]. Clean reads were mapped to the human genome GRCh38 by STAR [14] using the 2-pass model. Gene read counts were calculated using HTSeq-count [15]. GENCODE v22 GTF file was used to alignment and HTSeq-count. To quantify gene expression, the fragments per kilobase of transcript per million mapped reads (FPKM) values of each gene were calculated. To evaluate the RNA-seq data quality, multiple metrics include yield, alignment, GC bias, rRNA content, regions of alignment (exon, intron and intragenic), continuity of coverage, 3'/5' bias and count of detectable genes, among others were calculated by RNA-SeQC [16].
Snakemake workflow
To make this software extendable and scalable, I adopted snakemake [17,18] workflow engine to chain the tools, databases, and config files together. This enables users to easily make use of any cluster environment for processing and conveniently manage tools, databases and pipelines.
Results
I found that TOP200 was the best feature selection method after compared with other 4 methods mentioned above (Supplemental Table S1). 3817 genes were retained as feature genes (Supplemental Table S2). In the training phase, we performed 10-fold cross-validation on all training data to avoid overfitting and find best hyper-parameters. The accuracy, kappa, standard deviation of accuracy and standard deviation of kappa were 0.961, 0.959, 0.003292 and 0.003457, respectively. The median of sensitivity and specificity across 32 cancer types were 0.969 and 0.999, respectively (Supplemental Table S3). I used five external public RNA-seq datasets (Supplemental Table S4) to evaluate the performance of TRANSCUP. These datasets totally contain 557 samples and across 4 different cancer types including OSCC (HNSC), CRC, lung cancer (LUAD/LUSC) and BRCA. Notably, the overall accuracy of TRANSCUP was 98.7%, totally 7 samples were misclassified (Supplemental Table S5).
Conclusions
In this article, I described the TRANSCUP package for tumor type predication. TRANSCUP has been validated using 557 external samples and was more accurate than other methods for cancer type classification. Furthermore, TRANSCUP can analyze data from raw FASTQ to final cancer type predication results, and is more scalable and extendable than other methods. Users can train other kinds of models like deep learning models to extend its capability. However, this package needs to be validated on more external RNA-Seq datasets which including more diverse cancer types when data are available. Its actual clinical effect has to be verified by further experiments and clinical trials in the future.
Funding
None.
Conflict of Interest
None declared.
References
- Alshareeda AT, Al-Sowayan BS, Alkharji RR, et al. (2020) Cancer of unknown primary site: Real entity or misdiagnosed disease? J Cancer 11: 3919-3931.
- Rassy E, T Assi, N Pavlidis (2020) Exploring the biological hallmarks of cancer of unknown primary: Where do we stand today? Br J Cancer 122: 1124-1132.
- Pavlidis N, G Pentheroudakis (2012) Cancer of unknown primary site. Lancet 379: 1428-1435.
- Varadhachary GR, MN Raber (2014) Cancer of unknown primary site. N Engl J Med 371: 757-765.
- Overman MJ, Soifer HS, Schueneman AJ, et al. (2016) Performance and prognostic utility of the 92-gene assay in the molecular subclassification of ampullary adenocarcinoma. BMC Cancer 16: 668.
- Rosenfeld N, Aharonov R, Meiri E, et al. (2008) MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol 26: 462-469.
- Pillai R, Deeter R, Rigl CT, et al. (2011) Validation and reproducibility of a microarray-based gene expression test for tumor identification in formalin-fixed, paraffin-embedded specimens. J Mol Diagn 13: 48-56.
- Moran S, Martínez-Cardús A, Sayols S, et al. (2016) Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol 17: 1386-1395.
- Stark R, M Grzelak, J Hadfield (2019) RNA sequencing: the teenage years. Nat Rev Genet 20: 631-656.
- Flynn WF, Namburi S, Paisie CA, et al. (2018) Pan-cancer machine learning predictors of primary site of origin and molecular subtype. bioRxiv.
- Davalos V, M Esteller (2020) Insights from the genetic and transcriptional characterization of a cancer of unknown primary (CUP). EMBO Mol Med 12: e12685.
- Colaprico A, Silva TC, Olsen C, et al. (2016) TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44: e71.
- Bolger AM, M Lohse, B Usadel (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114-2120.
- Dobin A, Davis CA, Schlesinger F, et al. (2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: 15-21.
- Anders S, PT Pyl, W Huber (2015) HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166-169.
- DeLuca, DS, Levin JZ, Sivachenko A, et al. (2012) RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28: 1530-1532.
- Koster J, S Rahmann (2012) Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 28: 2520-2522.
- Singer J, Ruscheweyh HJ, Hofmann AL, et al. (2018) NGS-pipe: A flexible, easily extendable and highly configurable framework for NGS analysis. Bioinformatics 34: 107-108.
Corresponding Author
Peng Li, OrigiMed Inc., 5th Floor, Building 3, No.115 Xin Jun Huan Road, Minhang District, Shanghai-201114, China.
Copyright
© 2020 Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.