Genetic Factors Contributing to Alzheimer's Disease and FTLD
Abstract
Alzheimer's disease (AD) and frontotemporal lobar degeneration (FTLD) are two of the most prevalent neurodegenerative disorders, characterized by progressive cognitive decline and behavioral impairment. AD is the leading cause of dementia globally, while FTLD is the primary cause of early-onset dementia, particularly in individuals under 65. Despite clinical overlaps, these diseases differ in their pathological hallmarks, genetic underpinnings, and molecular mechanisms. This review aims to provide a brief overview of the genetic factors of AD and FTLD, highlighting their distinct pathogenesis in the progression of neurodegenerative diseases.
Keywords
Alzheimer's disease, FTLD, Neurodegenerative, Dementia, Pathological
Abbreviations
Aβ: Amyloid-Beta; ABCA7: ATP-Binding Cassette Sub-Family A Member 7; AD: Alzheimer’s Disease; APP: Amyloid Precursor Protein; APOE: Apolipoprotein E; BIN1: Bridging Integrator 1; CHMP2B: Charged Multivesicular Body Protein 2B; CLU: Clusterin; CR1: Complement Receptor 1; CSF: Cerebrospinal Fluid; EOAD: Early-Onset Alzheimer’s Disease; FAD: Familial Alzheimer’s Disease; FTD: Frontotemporal Dementia; FTLD: Frontotemporal Lobar Degeneration; FUS: Fused in Sarcoma; LOAD: Late-Onset Alzheimer’s Disease; MAPT: Microtubule-Associated Protein Tau; OPTN: Optineurin; PGRN: Progranulin; PICALM: Phosphatidylinositol-Binding Clathrin Assembly Protein; PSEN: Presenilin; SNPS: Single-Nucleotide Polymorphisms; SORL1: Sortilin Receptor 1; SQSTM1: Sequestosome 1; TBK1: TANK-Binding Kinase 1; TDP-43: TAR DNA-Binding Protein of 43 kDa; TREM2: Triggering Receptor Expressed on Myeloid Cells 2; VCP: Valosin-Containing Protein; VLDL: Very-Low-Density Lipoproteins
Introduction
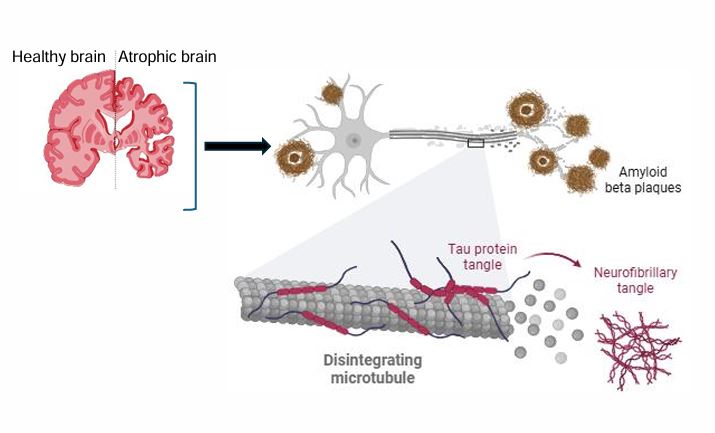
Neurodegenerative diseases represent a growing global health burden. Among them, AD and FTLD are two entities of major neurodegenerative disorders, leading to dementia, especially among young patients (<65-years-old) [1]. While both diseases result in neuronal loss and brain atrophy (Figure 1), they target different brain regions and involve divergent molecular pathologies [2]. AD primarily affects the hippocampus, entorhinal cortex, and temporal-parietal cortex, and FTLD predominantly involves the frontal and temporal lobes.
Alzheimer’s Disease (AD)
AD is a devastating neurodegenerative disorder and stands as the leading cause of dementia, accounting for an estimated 60-80% of all cases worldwide [3]. Currently, around 50 million individuals globally are grappling with some form of dementia. Alarmingly, as life expectancy continues to rise, projections suggest that by 2050, this figure could soar to 139 million, creating profound socioeconomic challenges and overwhelming our health systems [3].
AD progresses through stages, typically described as mild, moderate, and severe dementia, with mild cognitive impairment often preceding the dementia stages (Table 1). AD is typically classified by the age at which symptoms manifest. Early-onset Alzheimer’s disease (EOAD) usually strikes before the age of 60-65, while late-onset Alzheimer’s disease (LOAD) most commonly begins after 60. EOAD, sometimes referred to as familial Alzheimer’s disease (fAD), is linked to the inheritance of autosomal dominant mutations in three key genes: Amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). This form of the disease represents only about 5% of all AD cases. In contrast, the vast majority fall under the category of LOAD, which is also referred to as sporadic, a far more intricate disorder influenced by a complex interplay of genetic risk factors. While EOAD is unequivocally inherited, with a 100% heritability rate, LOAD is between 60-80% heritable, with environmental factors-such as diet, brain injury, lifestyle choices, and certain medications-contributing to the remaining cases [4].
Genes related to early-onset Alzheimer’s disease
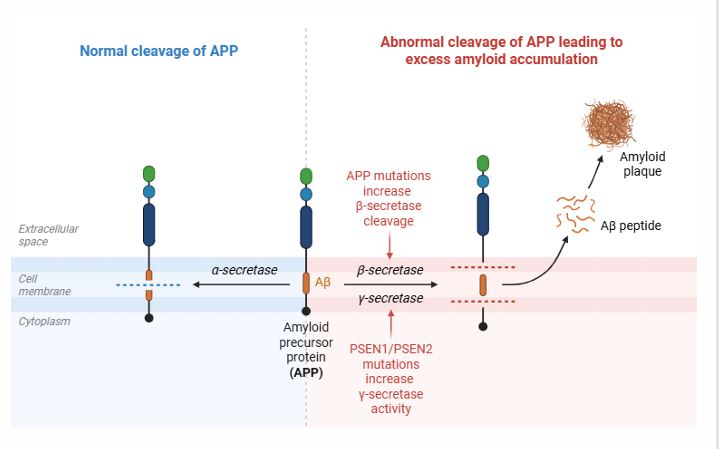
APP : APP is central to AD pathology, undergoing proteolytic processing through two main pathways: non-amyloidogenic and amyloidogenic. In the non-amyloidogenic route, APP is cleaved by α-secretase within the Aβ domain, which prevents Aβ formation and yields protective fragments. In contrast, the amyloidogenic pathway involves initial cleavage by β-secretase (BACE1), followed by γ-secretase, producing Aβ peptides (Aβ40 or Aβ42) (Figure 2) [5]. Of these, Aβ42 is more aggregation-prone, contributing to plaque formation in AD.
Non-Amyloidogenic Pathway (α-Secretase Pathway)-Neuroprotective
• APP is cleaved by α-secretase within the Aβ region, preventing the formation of Aβ.
• This generates soluble APPα (sAPPα) and a C-terminal fragment (C83).
• C83 is further processed by γ-secretase, producing a p3 peptide and an APP intracellular domain (AICD).
• Amyloidogenic Pathway (β-Secretase Pathway) – Aβ Production
• APP is first cleaved by β-secretase (BACE1), generating soluble APPβ (sAPPβ) and a C-terminal fragment (C99).
• C99 is then cleaved by γ-secretase (PSEN1/PSEN2/nicastrin/APH-1), releasing Aβ peptides (Aβ40 or Aβ42) and AICD.
• Aβ42 is prone to aggregation, forming oligomers, plaques, and contributing to AD pathology.
PSEN1 and PSEN2: Mutations in PSEN1 and PSEN2 cause fAD [6]. These mutations increase the production of neurotoxic Aβ42, which is more aggregation-prone than Aβ40 [7]. PSEN1 mutations are more common and lead to earlier onset (ages 30 to 50) and aggressive disease progression, while PSEN2 mutations are rarer [8].
Genes related to late-onset Alzheimer’s disease
Apolipoprotein E: Apolipoprotein E (APOE), a 34-kDa protein composed of 299 amino acids, is mainly produced by astrocytes, microglia, and choroid plexus cells within the central nervous system [9]. It exists in three primary isoforms: APOEε2, APOEε3, and APOEε4, differing at positions 112 and 158. These structural variations significantly impact APOE’s functions. APOEε2, which binds weakly to LDL receptors, offers a protective effect against AD. Conversely, APOEε4 is linked to reduced Aβ clearance and enhanced aggregation, substantially increasing AD risk depending on allele copy number.
APOE isoforms vary at positions 112 and 158: APOEε2 (Cys112, Cys158), APOEε3 (Cys112, Arg158), and APOEε4 (Arg112, Arg158) [10]. Changes in these amino acids alter APOE's structure. For example, Cys-158 in APOEε2 disrupts a salt-bridge, reducing positive potential and affecting receptor binding. Arg-112 in APOEε4 shifts lipid binding preference from HDL to very-low-density lipoproteins (VLDL). Additionally, the interaction between amino acids 61 and 112 affects lipoprotein binding, explaining APOEε4's higher affinity for VLDL compared to APOEε2 and APOEε3, which bind to HDL [11].
Clusterin: Clusterin (CLU), or apolipoprotein J (ApoJ), is a glycoprotein that plays a key role in AD pathology, particularly in lipid transport, protein folding, apoptosis, and inflammation [12]. It is linked to Aβ metabolism and clearance. Genome-wide association studies (GWAS) have identified CLU as a significant risk gene for late-onset AD, with certain polymorphisms increasing risk [12]. Specific polymorphisms in the CLU gene, particularly rs11136000 and rs9331888, have been associated with increased risk of AD [13]. CLU binds to Aβ peptides, influencing their aggregation, toxicity, and clearance, and facilitates Aβ transport across the blood-brain barrier, promoting microglial and astrocyte uptake for degradation.
In AD, CLU levels are elevated in the brain and cerebrospinal fluid (CSF) [14]. CLU is found in Aβ plaques and interacts with neurofibrillary tangles and modified Tau species. Lipidated CLU binds Aβ, forming complexes taken up by microglia via TREM2 [15]. This suggests that changes in CLU isoform expression may impact Aβ uptake and clearance.
Phosphatidylinositol-binding clathrin assembly protein: Phosphatidylinositol-binding clathrin assembly protein (PICALM) is crucial for clathrin-mediated endocytosis and is implicated in AD. PICALM facilitates the uptake of Aβ into microglia and endothelial cells for degradation. PICALM regulates the internalization of Aβ via low-density lipoprotein-receptor-related protein-1 (LRP1), promoting Aβ clearance and transcytosis [16]. Reduced PICALM levels impair Aβ clearance, leading to its accumulation, a hallmark of AD.
Bridging integrator 1: The Bridging Integrator 1 (BIN1) locus stands out as a pivotal genetic risk factor for late-onset AD [17]. Within the adult brain, BIN1 is predominantly expressed by oligodendrocytes, microglial cells, and glutamatergic neurons, with a striking reduction in expression noted in AD patients. This essential gene plays a vital role in membrane dynamics, endocytosis, cytoskeleton organization, and tau pathology, firmly linking it to neurodegeneration. While BIN1 is associated with both amyloid and tau pathology, some studies report contrasting findings, highlighting the complexity of this relationship [18]. A decrease in BIN1 expression has been shown to trigger an increase in Aβ production and accelerate tau aggregation in neurons [17]. Moreover, elevated levels of phosphorylated tau in the cerebrospinal fluid of AD patients have been found to correlate with genetic variants in the BIN1 locus, underscoring the intricate connection between genetics and the progression of Alzheimer's disease.
Complement receptor 1: Complement Receptor 1 (CR1) is a genetic risk factor for late-onset AD, identified in genome-wide association studies [19]. As a component of the complement system, CR1 is involved in immune response, synaptic pruning, and clearance of Aβ plaques. The single-nucleotide polymorphisms (SNPs) rs3818361 and rs6656401 in the CR1 gene are associated with an increased risk of AD [20,21]. Notably, rs6656401 has also been implicated in promoting vascular deposition of Aβ. Another study identified five SNPs (rs10494884, rs11118322, rs1323721, rs17259045, and rs41308433) linked to Aβ accumulation in the brain [22]. Notably, rs17259045 was associated with reduced Aβ accumulation in AD patients, while rs12567945 could increase CSF Aβ42 in the control population. CR1 mRNA levels correlate with neurofibrillary tangle density and tau abundance, suggesting CR1's role in inflammatory processes associated with AD. CR1's modulation of Aβ clearance, both peripherally via erythrocytes and directly in the brain by microglia, is crucial for its contribution to AD.
TREM2 : TREM2 (triggering receptor expressed on myeloid cells 2) is a key gene influencing the risk of LOAD [23]. It encodes a lipid receptor found in microglia, signaling through DAP12 to activate pathways that regulate microglial functions like phagocytosis and lipid processing. Mutations in the TREM2 gene are risk factors for Alzheimer's, similar to ApoE. TREM2 also interacts with several ligands, including ApoE and Aβ, which can activate TREM2 signaling [24]. The TREM2 R47H variant increases Alzheimer's disease risk by impairing TREM2 function and promoting neuritic dystrophy surrounding plaques [25]. Soluble TREM2 (sTREM2), generated through proteolytic cleavage of full-length TREM2, can be detected in the plasma and CSF of both Alzheimer's patients and healthy individuals [24]. Elevated levels of sTREM2 are seen early in the disease, correlating with brain amyloidosis and tau protein increases, and are associated with protective effects on microglial cell viability and enhanced clearance of amyloid plaques and damaged cells.
ATP-binding cassette sub-family A member 7: ATP-binding cassette sub-family A member 7 (ABCA7) is highly expressed in the brain, particularly in microglia, and plays a significant role in regulating phagocytosis [26]. Research shows that ABCA7 expression is low in astrocytes, moderate in neurons and oligodendrocytes, and high in microglia. Genetic variants in the ABCA7 locus are linked to an increased risk of AD, as ABCA7 is involved in lipid transport, Aβ peptide processing, and immune function. Patients with ABCA7 SNPs are more susceptible to AD, highlighting the importance of functional ABCA7 in protecting against the disease [27]. Furthermore, ABCA7 deficiency impairs clearance of Aβ and cytokine responses in immune cells. It acts as a protector by shuttling Aβ out of brain cells and potentially regulating its production, making it a promising target for AD therapies aimed at enhancing its activity.
Sortilin receptor 1 : Sortilin receptor 1 (SORL1) is crucial for the cellular trafficking of amyloid precursor protein (APP) and plays a significant role in generating Aβ peptides in AD [28]. It acts as a sorting receptor that regulates APP transport in neurons. Besides the APOEε4 allele, SORL1 is also genetically linked to late-onset AD. SORL1 helps trap APP in the Golgi apparatus, reducing Aβ production, which is associated with senile plaques in AD [29]. SORL1 underexpression leads to increased Aβ, while overexpression decreases APP and extracellular Aβ. Loss of SORL1 appears to be a causal factor for Alzheimer’s disease.
Frontotemporal Lobar Degeneration
FTLD includes a diverse group of disorders linked to behavioral changes, language impairment, and executive functioning deficits, caused by degeneration of the frontal and temporal lobes [30]. Frontotemporal dementia (FTD), affecting 15-22 per 100,000 individuals aged 45-65, is the second most common form of young-onset dementia [31]. Over the past two decades, research has identified various FTLD genes that explain most autosomal dominant cases, although sporadic cases remain less understood (Table 2). There is a growing need to identify additional FTLD risk genes, particularly with advancements in next-generation sequencing. The key genes associated with FTLD are presented below, highlighting their relevance to this condition.
Microtubule-associated protein tau
The Microtubule-Associated Protein Tau (MAPT) gene, which encodes the tau protein, is implicated in both AD and FTLD, particularly FTLD-Tau, a form of FTD characterized by tau protein inclusions [32]. MAPT encodes the tau protein, which stabilizes microtubules. Pathogenic mutations lead to tau hyperphosphorylation and aggregation, resulting in neuronal dysfunction and neurodegeneration. FTLD cases associated with tau pathology (FTLD-tau) account for approximately 40% of familial cases and are also implicated in other tauopathies such as progressive supranuclear palsy and corticobasal degeneration.
C9 or f72
C9 or f72 plays an essential role in multiple cellular processes, including autophagy, vesicle trafficking, lysosome maintenance, and mTORC1 regulation. Mutations in the C9 or F72 gene are the most common cause of familial ALS and FTLD identified to date [33]. The most common mutation in C9orf72 is a hexanucleotide repeat expansion (GGGGCC), which causes the formation of toxic RNA foci and the production of dipeptide repeat proteins (DPRs) through a non-canonical translation process [33]. These DPRs are believed to disrupt cellular functions, including RNA processing and protein homeostasis, contributing to TDP-43 aggregation and neurodegeneration [34].
GRN (Granulin)
Progranulin (PGRN) is a secreted protein that is taken up by cells and trafficked to the lysosome through receptors like Sortilin and Prosaposin [35]. Inside the lysosome, progranulin is cleaved by lysosomal enzymes (such as cathepsins) into seven granulin peptides, which are important for maintaining lysosomal function. Although initial characterization of PGRN function primarily focused on its role in extracellular signaling as a secreted protein, more recent studies revealed critical roles of granulin peptides in regulating lysosome function, including proteolysis and lipid degradation, consistent with its lysosomal localization [36]. Mutations in progranulin (PGRN) have been reported to be able to cause FTLD through haploinsufficiency, leading to accumulation of lipofuscin in lysosomal lumen, which impairs lysosomal function and increases neuroinflammation [37].
TARDBP (TDP-43)
TARDBP encodes TAR DNA-binding protein of 43 kDa (TDP-43), a protein involved in RNA splicing, transport, and stability [38]. Under normal conditions, TDP-43 is predominantly localized in the nucleus. Mutations in TARDBP cause TDP-43 to mislocalize to the cytoplasm, where it forms aggregates. In various models, mutations within the C-terminal domain of TDP-43 have been found to significantly enhance the intrinsic aggregation of this protein. Notably, the expression of specific TDP-43 mutations, including Q331K, M337V, Q343R, N345K, R361S, and N390D, has demonstrated a marked increase in aggregation and cell toxicity in yeast cells [39]. Moreover, other disease-associated mutations, such as G294A, Q331K, M337V, Q343R, N390D, and N390S, have similarly been shown to amplify protein aggregation when expressed in SH-SY5Y cells. These aggregates disrupt RNA metabolism and neuronal function, contributing to neurodegeneration in FTD and ALS. The phosphorylation of TDP-43 at serine residues 403/404 and 409/410 (p-TDP-43) plays a crucial role in the formation of the pathological inclusions characteristic of TDP-43 proteinopathies.
TANK-binding kinase 1
TANK-binding kinase 1 (TBK1) is a multifunctional serine/threonine kinase involved in inflammation, immune responses, and autophagy. TBK1 serves as a pivotal regulator of autophagy and mitophagy, orchestrating these vital processes through the phosphorylation of key autophagy cargo receptors, including Optineurin and Sequestosome 1 [40]. Mutations in TBK1 are associated with defective autophagy, which impairs the clearance of damaged proteins, including TDP-43 [41]. This leads to the accumulation of TDP-43 in the cytoplasm and contributes to neurodegeneration. TBK1 mutations are linked to both FTD and ALS, often presenting with a combination of cognitive decline and motor symptoms. TBK1 haploinsufficiency highlights the convergence of autophagy dysregulation in neurodegeneration [42].
Fused in Sarcoma
Fused in Sarcoma (FUS) is another RNA-binding protein involved in RNA transport and splicing [43]. Mutations in FUS result in cytoplasmic accumulation and aggregation of FUS protein. FUS proteins are predominantly localized to the nucleus, their mislocalization in the cytoplasm of the motor neurons can be observed in ALS patients, rare cases of FTLD-FUS have been reported, particularly in younger patients [44,45].
Valosin-containing protein
Valosin-Containing Protein (VCP) regulates protein degradation via the ubiquitin-proteasome system and autophagy [46]. Pathogenic mutations in VCP impair these pathways, leading to TDP-43 accumulation. Clinically, VCP mutations cause inclusion body myopathy, Paget disease of bone, and FTLD (collectively termed IBMPFD).
Charged Multivesicular Body Protein 2B
Charged Multivesicular Body Protein 2B (CHMP2B) is involved in endosomal-lysosomal trafficking via the ESCRT-III complex [47]. Mutations in CHMP2B disrupt endosomal sorting and autophagy, leading to ubiquitin-positive but TDP-43-negative inclusions, representing a rare pathological subtype of FTLD [48].
Optineurin and Sequestosome 1
Both Optineurin (OPTN) and Sequestosome 1 (SQSTM1) encode proteins that play a crucial role in autophagy and mitophagy [49,50]. Mutations disrupt autophagic clearance of misfolded proteins, contributing to TDP-43-positive inclusions in FTLD and ALS [51].
Involvement of Mitochondria in the Pathogenesis of AD and FTLD
Mitochondrial dysfunction and excessive production of reactive oxygen species (ROS) are pivotal contributors to the pathogenesis of AD and FTLD. Excess ROS production leads to widespread oxidative damage of lipids and proteins, which can contribute to the formation of lipofuscin, triggering lysosomal storage disease [52,53]. Oxidative modifications of proteins trigger a cascade of misfolding and aggregation of proteins such as Aβ, hyperphosphorylated tau, and TDP-43 [54,55]. These misfolded proteins accumulate into toxic aggregates, leading to the onset of neurodegenerative diseases. So, therapeutic targeting of dysfunctional mitochondrial clearance through the enhancement of mitophagy represents an effective approach to mitigate Aβ and tau pathology [56,57]. This strategy has been shown to reverse cognitive deficits in experimental models of Alzheimer’s disease.
Conclusion
Alzheimer's disease and FTLD represent distinct yet overlapping neurodegenerative disorders. While AD research has made significant strides, particularly in amyloid-targeting therapies, FTLD remains an urgent area for therapeutic innovation. Continued research into their molecular underpinnings, shared pathways, and divergent clinical manifestations will be essential for advancing early diagnosis, personalized medicine, and ultimately, disease prevention.
Data Availability
No data are associated with this article.
Funding Source
This study received no funding from outside sources.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Dubois B, Feldman HH, Jacova C, et al. (2014) Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol 13: 614-629.
- Chan D, Fox NC, Jenkins R, et al. (2001) Rates of global and regional cerebral atrophy in AD and frontotemporal dementia. Neurology 57: 1756-1763.
- Andrade Guerrero J, Santiago Balmaseda A, Jeronimo Aguilar P, et al. (2023) Alzheimer's disease: An updated overview of its genetics. Int J Mol Sci 24: 3754.
- Giri M, Zhang M, Lü Y (2016) Genes associated with Alzheimer's disease: An overview and current status. Clin Interv Aging 11: 665-681.
- Hampel H, Hardy J, Blennow K, et al. (2021) The amyloid-β pathway in Alzheimer's disease. Mol Psychiatry 26: 5481-5503.
- Petit D, Fernández SG, Zoltowska KM, et al. (2022) Aβ profiles generated by Alzheimer's disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol Psychiatry 27: 2821-2832.
- Arafi P, Devkota S, Williams E, et al. (2024) Alzheimer-mutant γ-secretase complexes stall amyloid β-peptide production. eLife 13: RP102274.
- Lin YS, Cheng CY, Liao YC, et al. (2020) Mutational analysis in familial Alzheimer's disease of Han Chinese in Taiwan with a predominant mutation PSEN1 p.Met146Ile. Sci Rep 10: 19769.
- Troutwine BR, Hamid L, Lysaker CR, et al. (2022) Apolipoprotein E and Alzheimer's disease. Acta Pharm Sin B 12: 496-510.
- Yamazaki Y, Zhao N, Caulfield TR, et al. (2019) Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat Rev Neurol 15: 501-518.
- McIntosh AM, Bennett C, Dickson D, et al. (012) The apolipoprotein E (APOE) gene appears functionally monomorphic in chimpanzees (Pan troglodytes). PLoS One 7: e47760.
- Foster EM, Dangla Valls A, Lovestone S, et al. (2019) Clusterin in Alzheimer’s disease: Mechanisms, genetics, and lessons from other pathologies. Front Neurosci 13: 164.
- Shuai P, Liu Y, Lu W, et al. (2015) Genetic associations of CLU rs9331888 polymorphism with Alzheimer's disease: A meta-analysis. Neurosci Lett 591: 160-165.
- Milinkeviciute G, Green KN (2023) Clusterin/apolipoprotein J, its isoforms and Alzheimer's disease. Front. Aging Neurosci 15: 1167886.
- Yeh FL, Wang Y, Tom I, et al. (2016) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 9: 328-340.
- Ando K, Nagaraj S, Küçükali F, et al. (2022) PICALM and Alzheimer's disease: An update and perspectives. Cells 11: 3994.
- Saha O, Melo de Farias AR, Pelletier A, et al. (2024) The Alzheimer's disease risk gene BIN1 regulates activity-dependent gene expression in human-induced glutamatergic neurons. Mol Psychiatry 29: 2634-2646.
- Voskobiynyk Y, Roth JR, Cochran JN, et al. (2020) Alzheimer's disease risk gene BIN1 induces Tau-dependent network hyperexcitability. Elife 9: e57354.
- Zhu XC, Yu JT, Jiang T, et al. (2015) CR1 in Alzheimer's disease. Mol Neurobiol 51: 753-765.
- Biffi A, Shulman JM, Jagiella JM, et al. (2012) Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology 78: 334-341.
- Thambisetty M, An Y, Nalls M, et al. (2013) Study of aging and the Alzheimer's disease neuroimaging initiative. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol Psychiatry 73: 422-428.
- Zhu XC, Dai WZ, Ma T (2020) Impacts of CR1 genetic variants on cerebrospinal fluid and neuroimaging biomarkers in alzheimer's disease. BMC Med Genet 21: 181.
- Li Y, Xu H, Wang H, et al. (2023) TREM2: Potential therapeutic targeting of microglia for Alzheimer's disease. Biomed Pharmacother 165: 115218.
- Qin Q, Teng Z, Liu C, et al. (2021) TREM2, microglia, and Alzheimer's disease. Mech Ageing Dev 195: 111438.
- Cheng Hathaway PJ, Reed Geaghan EG, Jay TR, et al. (2018) The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer's disease. Mol Neurodegener 13: 29.
- Hongyun Li, Tim Karl, Brett Garner (2015) Understanding the function of ABCA7 in Alzheimer's disease. Biochem Soc Trans 43: 920-923.
- Santos García I, Bascuñana P, Brackhan M, et al. (2025) The ABC transporter A7 modulates neuroinflammation via NLRP3 inflammasome in Alzheimer's disease mice. Alzheimers Res Ther 17: 30.
- Huang CC, Liu ME, Kao HW, et al. (2016) Effect of Alzheimer's disease risk variant rs3824968 at SORL1 on regional gray matter volume and age-related interaction in adult lifespan. Sci Rep 6: 23362.
- Andersen OM, Schmidt V, Spoelgen R, et al. (2006) Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45: 2618-2628.
- Pottier C, Ravenscroft TA, Sanchez Contreras M, et al. (2016) Genetics of FTLD: Overview and what else we can expect from genetic studies. J Neurochem 1: 32-53.
- Onyike CU, Diehl Schmid J (2013) The epidemiology of frontotemporal dementia. Int Rev Psychiatry 25: 130-137.
- Strang KH, Golde TE, Giasson BI (2019) MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest 99: 912-928.
- Licata NV, Cristofani R, Salomonsson S, et al. (2022) C9orf72 ALS/FTD dipeptide repeat protein levels are reduced by small molecules that inhibit PKA or enhance protein degradation. EMBO J 41: e105026.
- Cook CN, Wu Y, Odeh HM, et al. (2020) C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med 12: eabb3774.
- Du H, Zhou X, Feng T, et al. (2022) Regulation of lysosomal trafficking of progranulin by sortilin and prosaposin. Brain Commun 4: fcab310.
- Root J, Mendsaikhan A, Taylor G, et al. (2024) Granulins rescue inflammation, lysosome dysfunction, lipofuscin, and neuropathology in a mouse model of progranulin deficiency. Cell Rep 43: 114985.
- Simon MJ, Logan T, DeVos SL, et al. (20230 Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell Biol 33: 324-339.
- Hu Y, Hruscha A, Pan C, et al. (2024) Mis-localization of endogenous TDP-43 leads to ALS-like early-stage metabolic dysfunction and progressive motor deficits. Mol Neurodegener 19: 50.
- Jo M, Lee S, Jeon YM, et al. (2020) The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp Mol Med 52: 1652-1662.
- Gurfinkel Y, Polain N, Sonar K, et al. (2022) Functional and structural consequences of TBK1 missense variants in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurobiol Dis 174: 105859.
- Harding O, Evans CS, Ye J, et al. (2021) ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc Natl Acad Sci USA 118: e2025053118.
- Lamb R, Rohrer JD, Real R, et al. (2019) A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Cold Spring Harb Mol Case Stud 5: a003913.
- Hou J, Yang S, Guo Y, et al. (2023) FUS regulates the alternative splicing of cell proliferation genes related to atherosclerosis. Exp Biol Med (Maywood) 248:1459-1468.
- Rezvykh AP, Ustyugov AA, Chaprov KD, et al. (2023) Cytoplasmic aggregation of mutant FUS causes multistep RNA splicing perturbations in the course of motor neuron pathology. Nucleic Acids Res 51: 5810-5830.
- Goodwill V, Coughlin D, Pizzo D, et al. (2021) A case of frontotemporal lobar degeneration with fus-positive pathology (ftld-fet) with corticobasal features and language deficits. J Neuropathol Exp Neurol 80: 890-892.
- Wani A, Zhu J, Ulrich JD, et al. (2021) Neuronal VCP loss of function recapitulates FTLD-TDP pathology. Cell Rep 36: 109399.
- Cheruiyot A, Lee JA, Gao FB, et al. (2014) Expression of mutant CHMP2B, an ESCRT-III component involved in frontotemporal dementia, causes eye deformities due to Notch misregulation in Drosophila. FASEB J 28: 667-675.
- Deng X, Sun X, Yue W, et al. (2022) CHMP2B regulates TDP-43 phosphorylation and cytotoxicity independent of autophagy via CK1. J Cell Biol 221: e202103033.
- Lee KW, Ryu KJ, Kim M, et al. (2024) RCHY1 and OPTN are required for melanophagy, selective autophagy of melanosomes. Proc Natl Acad Sci USA 121: e2318039121.
- Kumar AV, Mills J, Lapierre LR (2022) Selective autophagy receptor p62/SQSTM1, a pivotal player in stress and aging. Front Cell Dev Biol 10: 793328.
- Ho PC, Hsieh TC, Tsai KJ (2024) TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: From pathomechanisms to therapeutic strategies. Ageing Res Rev 100: 102441.
- Klemmensen MM, Borrowman SH, Pearce C, et al. (2024) Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics 21: e00292.
- Sikder MM, Li X, Akumwami S, et al. (2025) Reactive oxygen species: Role in pathophysiology, and mechanism of endogenous and dietary antioxidants during oxidative stress. Chonnam Med J 61: 32-45.
- Bhatt S, Puli L, Patil CR (2021) Role of reactive oxygen species in the progression of Alzheimer's disease. Drug Discov Today 26: 794-803.
- Zuo X, Zhou J, Li Y, et al. (2021) TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat Struct Mol Biol 28: 132-142.
- Jia N, Ganesan D, Guan H, et al. (2025 ) Mitochondrial bioenergetics stimulates autophagy for pathological MAPT/Tau clearance in tauopathy neurons. Autophagy 21: 54-79.
- Fang EF, Hou Y, Palikaras K, et al. (2019) Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci 22: 401-412.
Corresponding Author
Mohammad Mamun Sikder, Department of Molecular Biology and Genetics, Weill Institute for Cell and Molecular Biology, Cornell University, 345 Weill Hall, Ithaca, NY 14853, USA, Tel: +1-607-339-7578, Fax: +1-607-2555961.
Copyright
© 2025 Sikder MM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.