Aquaporin-4 Inhibition Mediates Piroxicam-Induced Neuroprotection against Focal Cerebral Ischemia/Reperfusion Injury in Rodents
Abstract
Background
Brain edema due to the over expression of AQP4 is a major determinant for progression of neuronal insult during ischemic stroke. During brain pathology, both down regulation and up regulation of AQP4 expression is observed. However, AQP4 over expression is one of the main contributors of water imbalance occurring in the brain during ischemic stroke.
Objectives
In our previous studies we have shown that binding of Piroxicam to AQP4 takes place with an optimal binding energy. Following which, we hypothesized that Piroxicam in ischemic stroke may be neuroprotective. This neuroprotective effect is rendered via regulation of AQP4 which mitigates cerebral ischemia.
Methods
Rats were treated with placebo OR Piroxicam at 30 min prior, 2 h post and 4 h post MCAO (60 minutes), followed by reperfusion for 24 hours. Animals were then evaluated for motor function and neurological deficits just prior to sacrifice. Brains were harvested for infarct volume assessment, biochemical analysis, water content measurement, calcium estimation, RT-PCR and western blotting.
Results
Pretreatment with Piroxicam thirty minutes prior to ischemia and four hour post reperfusion conferred neuroprotection as observed through a significant improvement in motor behavior and neurological deficit, decrease in cerebral infarct volume and brain edema. This neuroprotection was found to be concomitant with the down regulated AQP4 expression along with the inhibition of acid mediated rise in intracellular calcium levels.
Conclusion
Results from the present study offer substantial evidence that Piroxicam confers neuroprotection in focal cerebral ischemia by acting as a potent AQP4 modulator.
Keywords
Cerebral ischemia, Aquaporin-4, Brain edema, Piroxicam, Neuroprotection
Abbreviations
MDA: Malondialdehyde; NO: Nitric Oxide; AQP4: Aquaporin-4; NSAID: Non-steroidal anti-inflammatory drug; COX-2: Cyclooxygenase-2
Introduction
Cerebral edema is a devastating complication of various neurological disorders that accounts for mortality and morbidity [1-3]. Several other mechanisms exacerbate the neuronal deterioration. Therapeutic options for treating cerebral edema are highly limited, with none of those being able to successfully obliterate the molecular mechanisms responsible for edema. This necessitates and suggests the use of pharmacological molecules which could intervene cerebral edema [2,4].
Aquaporin-4 (AQP4) are integral membrane proteins, playing an important role in maintaining water homeostasis in the central nervous system and its dysfunction may lead to brain edema [3]. AQP4 is a bidirectional water channel playing an important role in maintaining brain water homeostasis [3] .This protein is expressed robustly in the BBB and CSF-brain interfaces astroglia [5], involved in water transfer between brain parenchyma and fluid compartments (blood and CSF). It is suggested that deletion of AQP4 markedly reduced brain swelling of cytotoxic brain edema, including focal ischemia [6,7]. Levels of AQP4 are distinctly altered in experimental models of swelling and brain injury in response to neuronal ischemic insult [2,8]. When subjected to MCAO, AQP4 deficient mice show better functional and neurological outcome as compared to control mice [7]. Since AQP4 seems to facilitate movement of water in cytotoxic edema, expression level of AQP4 can indirectly define the extent of brain swelling following cerebral ischemia. Therefore, targeting AQP4 by pharmacological molecule may represent a potential therapeutic agent for treating brain edema [2].
One of the prominent events taking place following a pathological condition like stroke is the free radical mediated injury. Free radicals play a critical role in brain damage following ischemia by exacerbating membrane damage, ultimately leading to neuronal cell death. Several therapeutic strategies have been reported in past studies which have shown to reduce damage mediated by free radical following ischemic stroke [9-12]. Ultimate result of cerebral ischemia is brain injury, which is associated with neurobehavioral and neurological deficits that depend on the region or part of the brain or networks that are disrupted [13]. Hence, the need of the hour is to identify pharmacological molecules which can act in a multifaceted dimension.
Prior research have showed that non-steroidal anti-inflammatory drugs (NSAIDs) inhibit acidotoxicity and inflammation by acting against acid sensing ion channels and inflammatory mediators. However, but effects on AQP4 and cognitive function by NSAID have not been reported till date to the best of our knowledge [14]. Previous in-silico studies from our lab have shown that Piroxicam may be one of the molecules of choice to reduce acidotoxicity mediated by acid sensing ion channel 1a (ASIC1a), edema resulting from brain stroke, matrix metalloproteinases and µ-calpain inhibition mediated neuroprotection [15] and cognitive deficits mediated by stroke concomitantly [16,17]. Hence, the present study has been performed with Piroxicam as a candidate NSAID, whose efficacy as a neuroprotective agent is yet to be explored in vivo targeting the expression of AQP4. In vitro neuroprotective effects of Piroxicam have been demonstrated in neuronal cells [18,19]. For this study, we have tried to determine the neuroprotective efficacy of Piroxicam in a rodent model of focal cerebral ischemia and have also explored its neuroprotective effects with AQP 4 channel as one of the targets to inhibit stroke mediated brain edema.
Material and Methods
Chemicals
Most of the chemicals were obtained from Sigma Aldrich unless otherwise mentioned. Antibodies were also procured Molecular biology grade chemicals and enzymes were used as per manufacturer's instructions.
Animals
Male Charles Foster rats (6 weeks, 250 ± 10 g) were received from Central Animal House of Banaras Hindu University (Reg No.- 542/AB/CPCSEA; D/09-10/765). Animals were kept under standard laboratory conditions with the highest standards of care and housing. Animals were provided with commercial diet having standard ingredients. Animals were fasted overnight to control glucose level with free access to water and maintained at 12 h day/night cycle. Approved procedures and the institutional animal ethical committee guidelines were followed throughout the study.
Focal cerebral ischemia
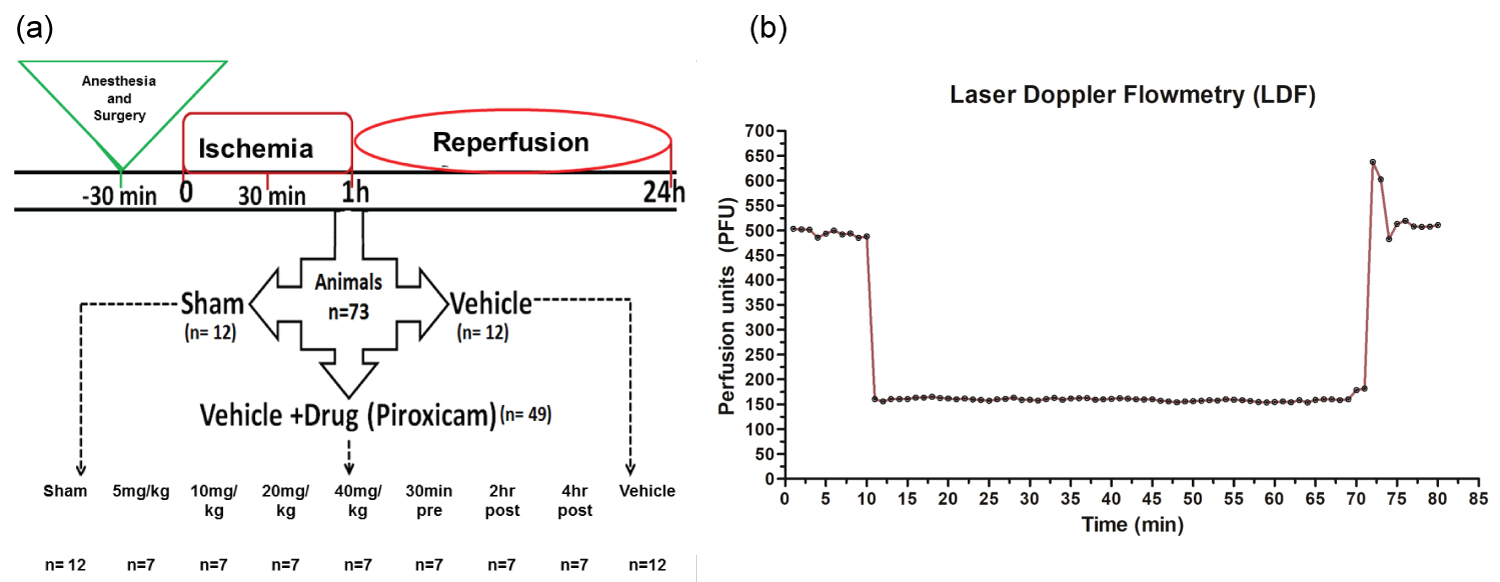
Focal cerebral ischemia was induced by occlusion of middle cerebral artery (MCA) with modified intraluminal technique [20]. Rats were anesthetized by a combination of ketamine (75 mg/ kg b.w.) and xylazine (10 mg/kg b.w.) and then put on the surgical table with a heating pad to maintain body temperature of 37 ± 0.5 ℃. Reperfusion for 24 hrs was done by gently withdrawing the filament after 60 min of ischemia. Animals were allowed to recover from anesthesia with temperature maintained at 26 ± 2.5 ℃ with food and water supply in ad-libitum. In sham group, all procedures were carried on except the filament insertion. Brain doppler recording confirmed that all rats with MCAO, had blood flow reduced by approximately 70% of pre-ischemic values within 5-7 mins of filament insertion (Figure 1). Rats not exhibiting significant drop in cerebral blood flow were excluded from the study.
Dose optimization of Piroxicam and experimental design
Piroxicam was administered i.p in four doses of 5 mg, 10 mg, 20 mg and 40 mg/kg b/w. to find the efficacious dose. Piroxicam in optimized dose was dissolved in normal saline and were put to physiological pH before i.p. administration 30 min before inducing focal cerebral ischemia and 2hr and 4hr post ischemia. 73 animals were divided into six groups consisting of sham (n = 12), vehicle (n = 12) and Piroxicam treated (n = 7) for each dose group. Further, biochemical and molecular studies were conducted with the effective dose in three more groups viz. sham, vehicle (n = 12 in each group) and treated (n = 7). All experiments were conducted in the morning. Room temperature and humidity were kept constant. The experimenter was blinded to the treatments to rats. Representation of the experimental design is in Figure 1. The total animals reported are less considering of the co-morbidity factors. Rectal temperature of the rats was measured at an ambient temperature of 21.5 ± 1 ℃ Temperature was recorded before any drug treatment and thereafter every 60 min up to 8 h. Arterial blood parameters (pH, PaCO2, PaO2) and MAP were monitored throughout the experiment which underwent MCAO and drug treatment, but no significant differences between the experimental groups were observed as shown in Table 1.
Evaluation of motor coordination and neurological scoring
Motor coordination was evaluated by testing the ability of rats to remain on revolving rod in a rotarod [21]. The rate of rotation was adjusted in such a manner that it allowed the normal rats to stay on it for five minutes. Each rat was given five trials for 2 days before the actual reading was taken. The readings were taken at 10, 15, 20 and 25 rpm after 24 hr, 48 hr and 72 hr post MCAO [22]. An evaluation of the functional outcome was performed 24 h post reperfusion [20]. Neuro scores were derived on five point scale with total of 10 grading scores; 0 for no neurological deficit; 1 for inability to extend opposite forepaw, 2 for contralateral circling, 3 for inability to grip the wire meshes and 4 for depressed level of consciousness. Rats not exhibiting neurological score, having small infarct sizes and inconsistent physiological parameters were excluded from the study.
Estimation of cerebral infract volume and brain swelling
After neurological examination, rats were sacrificed and coronal brain sections (1 mm thick) for histology were obtained. Infarct area were measured and quantified by image analysis software (NIH image J). Further, infarct volume was calculated in mm3 [23]. Brain swelling was calculated according to the following formula [24]:
Biochemical analysis
Biochemical analysis comprised of measuring nitrite, malondialdehyde and calcium influx, 20 min and 60 min post ischemia respectively in cortex and striatal regions. Animals were decapitated and brain areas were quickly removed and homogenized (5:1 v/w) in ice cold 0.1 M phosphate buffer, pH 7.4. The tissue homogenate thus obtained was used for estimation as per reported protocol given below.
Measurement of nitrite
Nitrite and nitrate are generally the markers for NO production in a damaged cell or tissue. The nitrite levels were estimated in affected brain regions using Griess reaction [25]. Nitrite was measured as per reported studies [23,26].
Estimation of malondialdehyde (MDA)
The MDA is a by-product of lipid peroxidation used as a a biomarker for membrane damage. It was determined based on its reaction with thiobarbituric acid (TBA). MDA was measured as per reported studies [23]. The colour formed was measured at 532 nm in spectrophotometer [27].
Estimation of Ca2+ influx
The levels of Ca2+ in cortical and striatal region of all groups were estimated by using Fura 2AM with minor modification of previously mentioned method of Pokorski, et al. [28]. The ratio between the fluorescent intensities of Fura 2-Ca2+ complex and the unchealated Fura 2 (F340/F380) reflected the Ca2+ concentration.
Molecular Studies
Studies reported maximum and stable AQP4 expression at 24 hr post ischemia, hence we performed molecular studies 1/24 hr post stroke [29-31].
Isolation of total RNA
Expression of the gene for AQP4 in the striatum and cortex were studied by semi-quantitative RT-PCR using a MJ Research Thermal Cycler. Total RNA was isolated using TRI reagent (Sigma) as per user manual and dissolved in DEPC-treated water. DNase I (DNA free ambion) digestion of total RNA was done according to manufacturer's guideline to remove any DNA contamination. The RNA was quantified by spectrophotometry [30,31].
Reverse Transcription
The first strand of cDNA was synthesized from 2.0 µg of total RNA, by using random hexamer primer and MuLV reverse transcriptase (Revert AidTM H Minus, 200 units, MBI Fermentas). RT was performed as per previous reported studies [30,31].
Polymerase Chain Reaction
PCR was performed as per previous reported studies [30,31]. For PCR, 10 ng of cDNA and 0.2 µM of primers for AQP4 and β-actin were used. Expression of AQP4 and β-actin was assessed by polymerase chain reaction (PCR) using the following gene-specific primers: 5'GGAAGGCTAGGTTGGTGACTTC3' and 5'TGGTGACTCCCAATCCTCCAAC 3' for AQP4; 5'ATCGTGGGCCGCTCTAGGCACC 3' and 5'CTCTTTGATGTCACGCACGATTTC 3' for β-actin [30,31]. The resulting gel bands were visualized in a UV-transilluminator and photographed by Nikon digital camera. The relative amount of the AQP4 was expressed as optical density relative to that of the β-actin [30,31].
Tissue lysate preparation
Cortex and striatum were homogenized in 50 mM Tris-Cl, (pH 7.4), having 0.2% triton X-100, 5 mM EDTA, 5 mM EGTA, 2 mM PMSF, 5 mM benzamidine, 2 mM b-ME, and protease inhibitor cocktail (Sigma-Aldrich). Protein was estimated by Bradford method using BSA as standard [30-32].
SDS-Polyacrylamide gel electrophoresis
For Western blotting, 50 µg of crude sample was denatured in 5X Laemelli gel loading buffer (100 mM Tris-Cl (pH 6.8), 2% sodium dodecyl sulfate, 2% b-ME, 20% glycerol and 0.2% bromophenol blue) in boiling water bath for 5 min. Methods followed as per previous reported studies. For proper stacking, the samples were run at 15 mA in stacking gel and resolved at 30 mA in resolving gel [30,31].
Immunoblotting
Immunoblotting for AQP4 (Sigma, A5971, 1:1000) was performed as per reported protocol [30-32]. β-actin (Sigma, A2228, 0.5:1000) was used as control for immunoblotting. Band density values were normalized to β-actin [30-32].
Protein estimation
Protein content was estimated by the method of Bradford using bovine serum albumin as standard [30-32].
Statistical Analysis
The mean ± SEM was analyzed by Origin software. All the data were examined by one-way ANOVA followed by Student-Newman-Keuls test. P < 0.05 and < 0.01 was taken as statistically significant. For RT-PCR, the signal intensity of the target band, and for immunoblots, monomeric as well as higher oligomeric bands were measured after normalization with β-actin and expressed as relative densitometry value (RDV).
Results
Dose optimization of Piroxicam
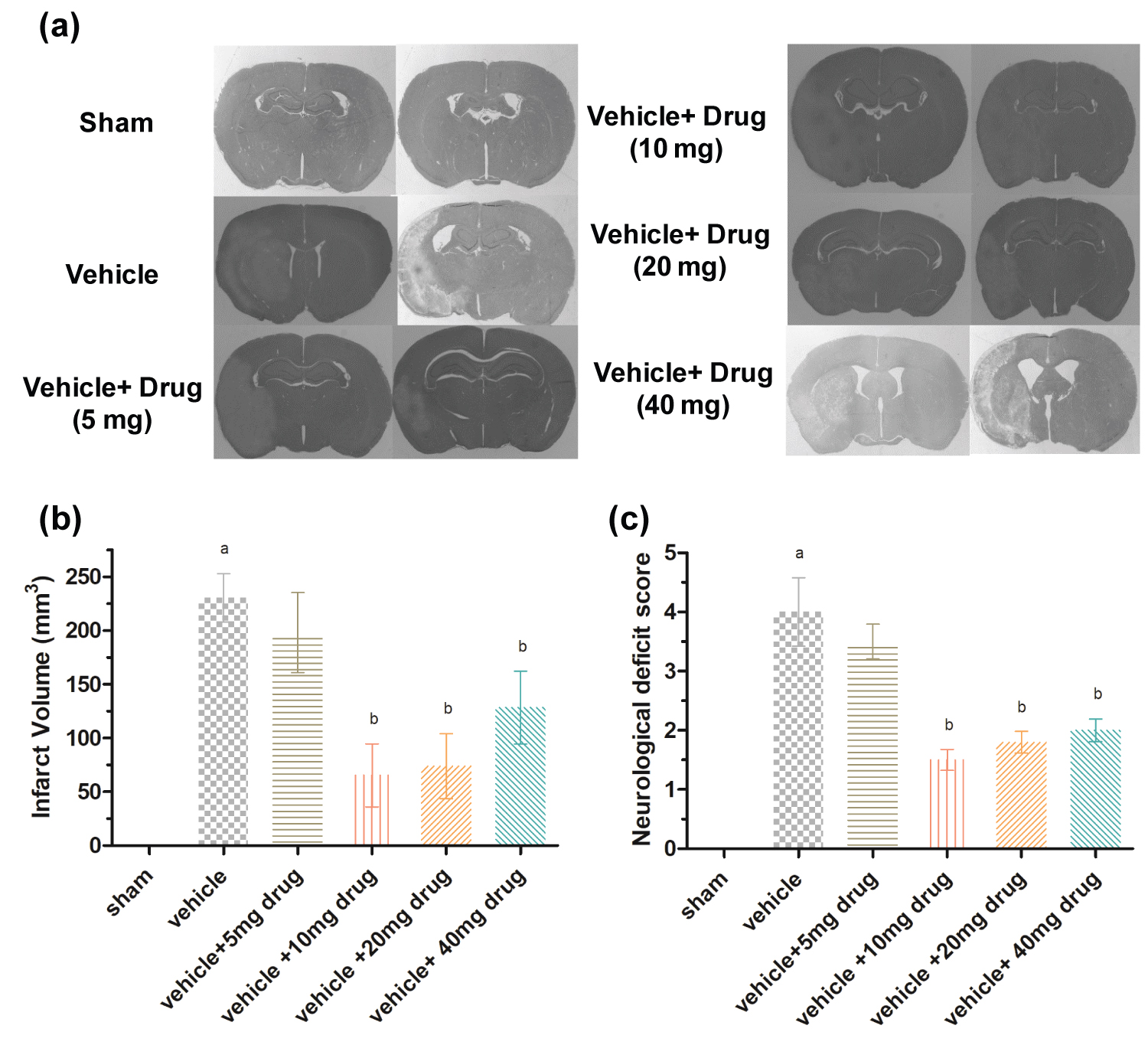
Minimum neuroprotective dose of Piroxicam was determined on the basis of improvement in functional outcome and reduced cerebral infarct volume of rats subjected to ischemic stroke. Marked infarcts were obtained in both cortical and striatal regions of rat brain as observed in histology coronal brain sections (Figure 2a). The mean of infarct volume was found to be 230.32 ± 19.32 mm3 in vehicle treated control rats whereas pretreatment with Piroxicam at 5, 10, 20, 40 mg/Kg i.p. doses produced marked decrease in infarct volume, ranging from 198 ± 32.2, 65.36 ± 25.3, 73.87 ± 26.2, 128.37 ± 29.4 mm3 respectively (P ≤ 0.05 ) (Figure 2b). Neuroscores were obtained post ischemia in all groups. The vehicle group had significant higher neurological deficit as compared to sham group while significant (P < 0.05) improvement in neurological deficit score was found in Piroxicam pretreated rats at all doses except lowest dose as compared to vehicle treated group (Figure 2c). Thus 10 mg/kg Piroxicam i.p. showed significant (P < 0.01) improvement in neurological deficit and infarct size.
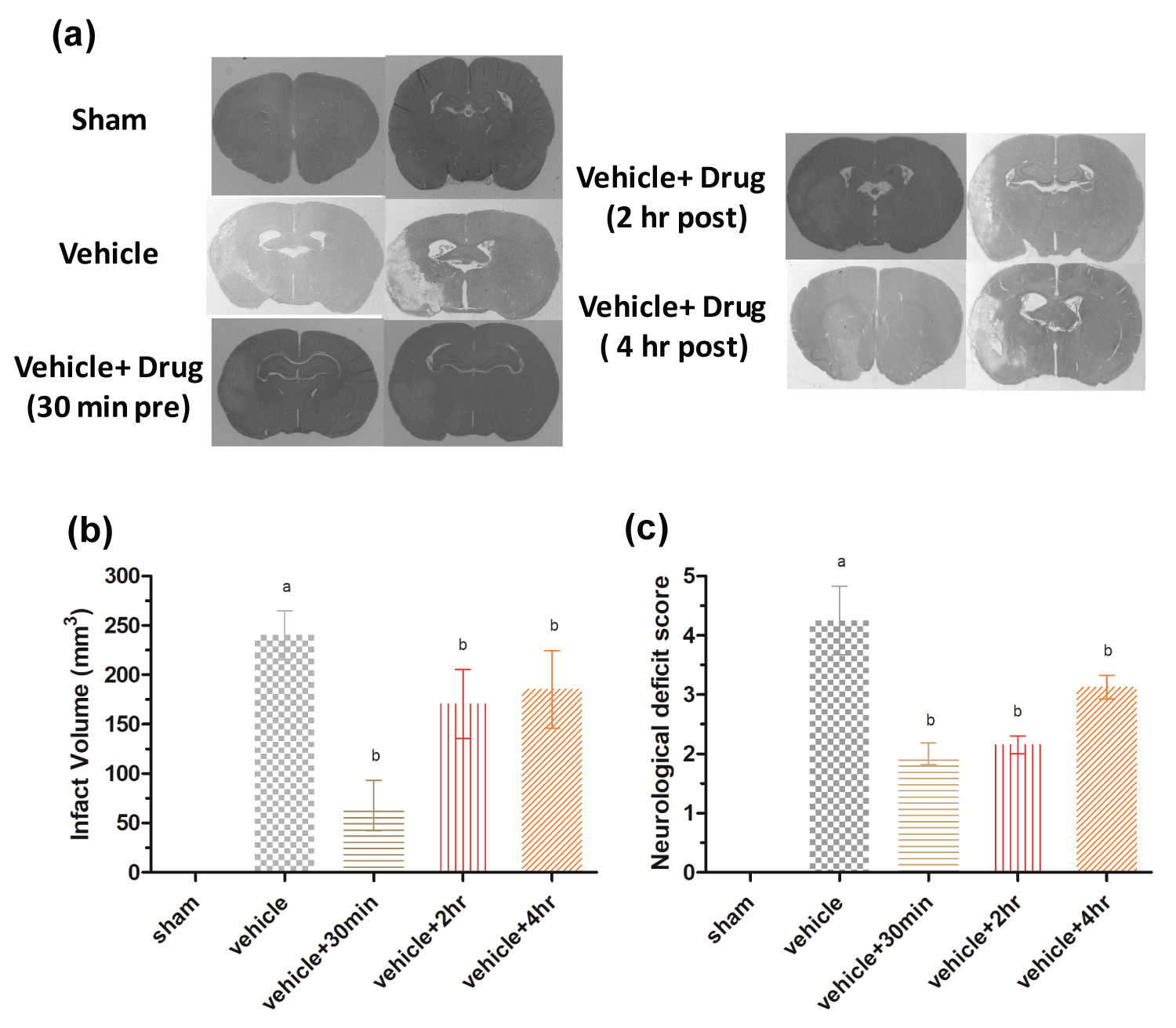
Effect of Piroxicam on pre and post treatment on cerebral infarct volume and neurological deficit
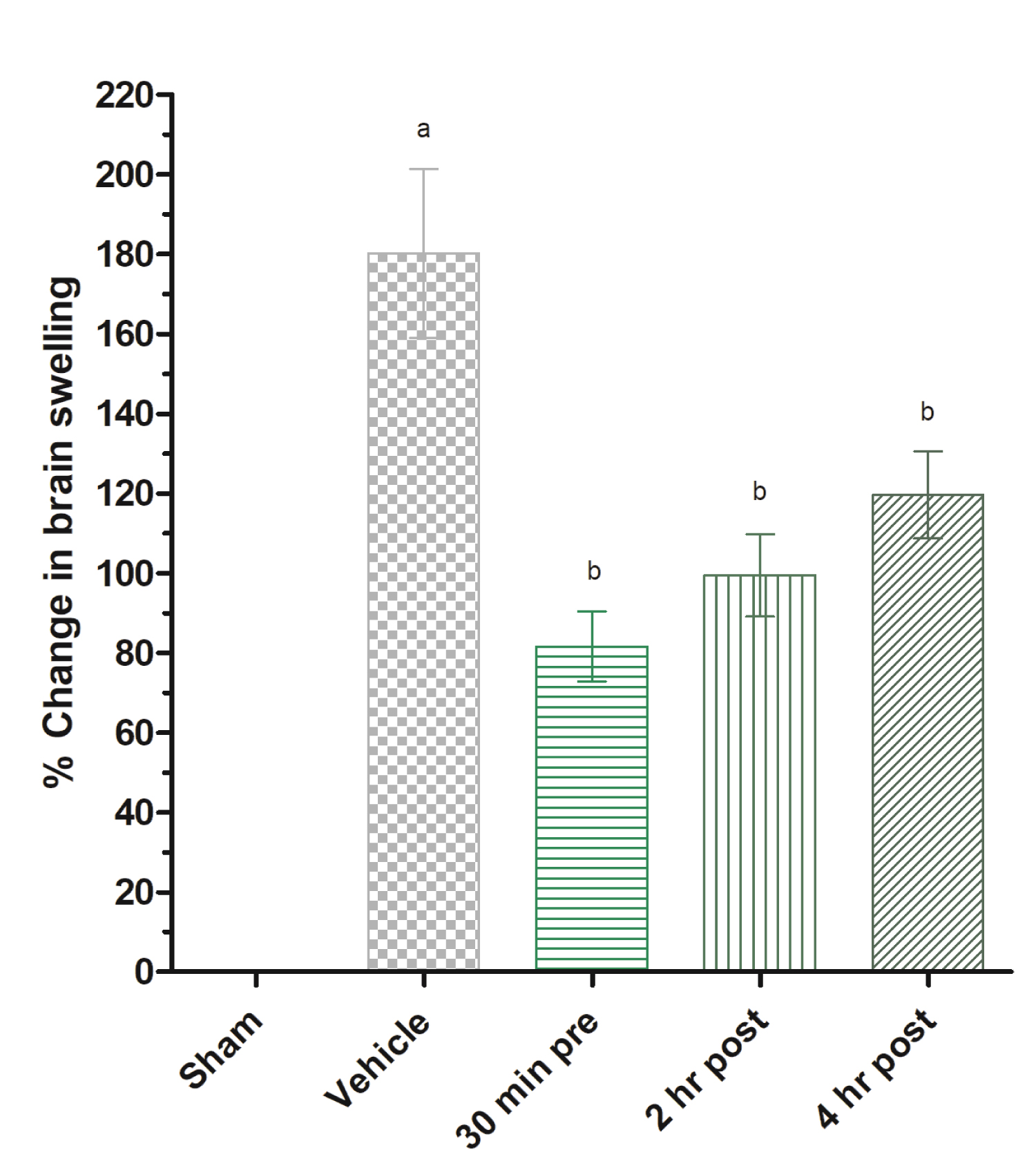
The mean of infarct volume was found to be 240.34 ± 21.3 mm3 in vehicle treated control rats while in Piroxicam treatment group at 30 min pre, 2 h post and 4 h post produced marked decrease in infarct volume, ranging from 67.78 ± 22.2, 170.37 ± 30.4, 185.36 ± 33.9, mm3 respectively (Figure 3a and Figure 3b). Neuroscores were obtained after stroke in all experimental groups. The vehicle group shows significantly more neurodeficit compared to sham group while significant (P < 0.05) improvement in functional outcome was observed in 10 mg/kg b.w Piroxicam administered rats at all doses except lowest dose as compared to vehicle treated group (Figure 3c). Thus 30 min Piroxicam pretreatment group showed significant (P < 0.05) improvement in neurodeficit and infarct volume. Brain swelling after ischemic stroke lead to increase in brain water content. This may be due to differential expression of brain water channels. Piroxicam 30 min pretreatment significantly decreased brain swelling as compared to vehicle (Figure 4).
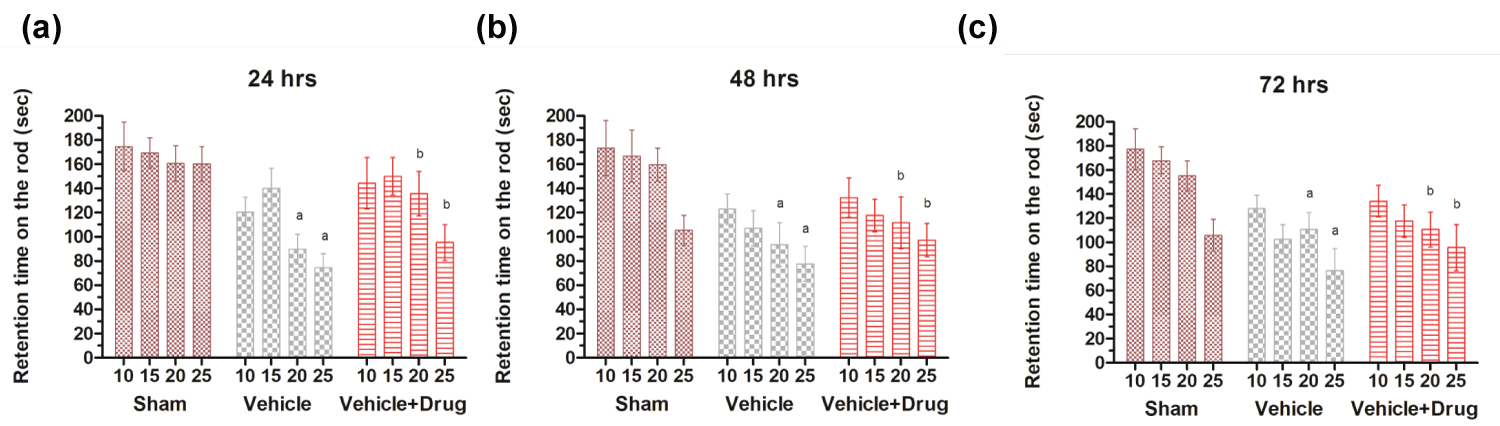
Analysis of rota rod test
Rotarod experiment showed a significant decrease in the retention time on the rotating rod in the vehicle and vehicle + drug administered rats at 10, 15, 20 and 25 revolutions per minute (rpm) when compared to sham. Piroxicam treatment in ischemic rats significantly (p < 0.05) reversed the retention time near to control at 10, 15, 20 and 25 rpm (Figure 5a, Figure 5b and Figure 5c). Long term outcomes up to 72 hrs were assessed in view to consider the STAIR criteria.
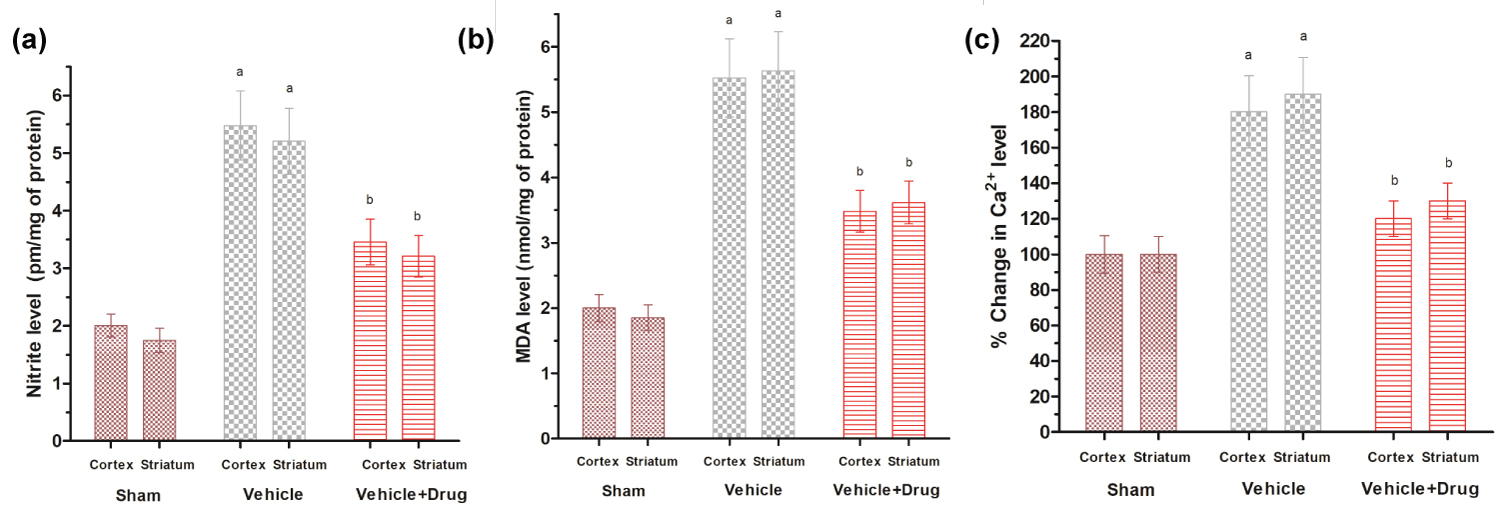
Effect of Piroxicam on brain nitrite levels
Nitrite levels were measured at 20 minutes post ischemia in cortex and striatal regions of rat brain as per previous reported study [23]. Piroxicam significantly (P < 0.05) attenuated nitrite levels in both striatal and cortical brain regions of rat brain (Figure 6a).
Effect of Piroxicam on brain MDA levels
MDA to check degree of lipid peroxidation was measured post 60 min of ischemia in cortex and striatal regions of rat brain [23]. The MDA levels in vehicle treated rats were found to be 5.48 ± 0.32 & 5.58 ± 0.82 nmol/mg proteins whereas Piroxicam i.p. treated rats were 3.32 ± 0.52 & 3.46 ± 0.75 nmol/mg proteins, in cortical and striatal regions, respectively (Figure 6b). Thus Piroxicam pretreatment significantly (P < 0.05) reduced MDA in post ischemic brain.
Effect of Piroxicam on Ca2+ level
One of the major outcomes of stroke is robust Ca2+ influx. Treatment of Piroxicam inhibited increase in Ca2+ in cortex and striatum. A significant inhibition of calcium was observed in Piroxicam treated group which advocate the calcium channel blocking capacity of Piroxicam (P ≤ 0.05) (Figure 6c).
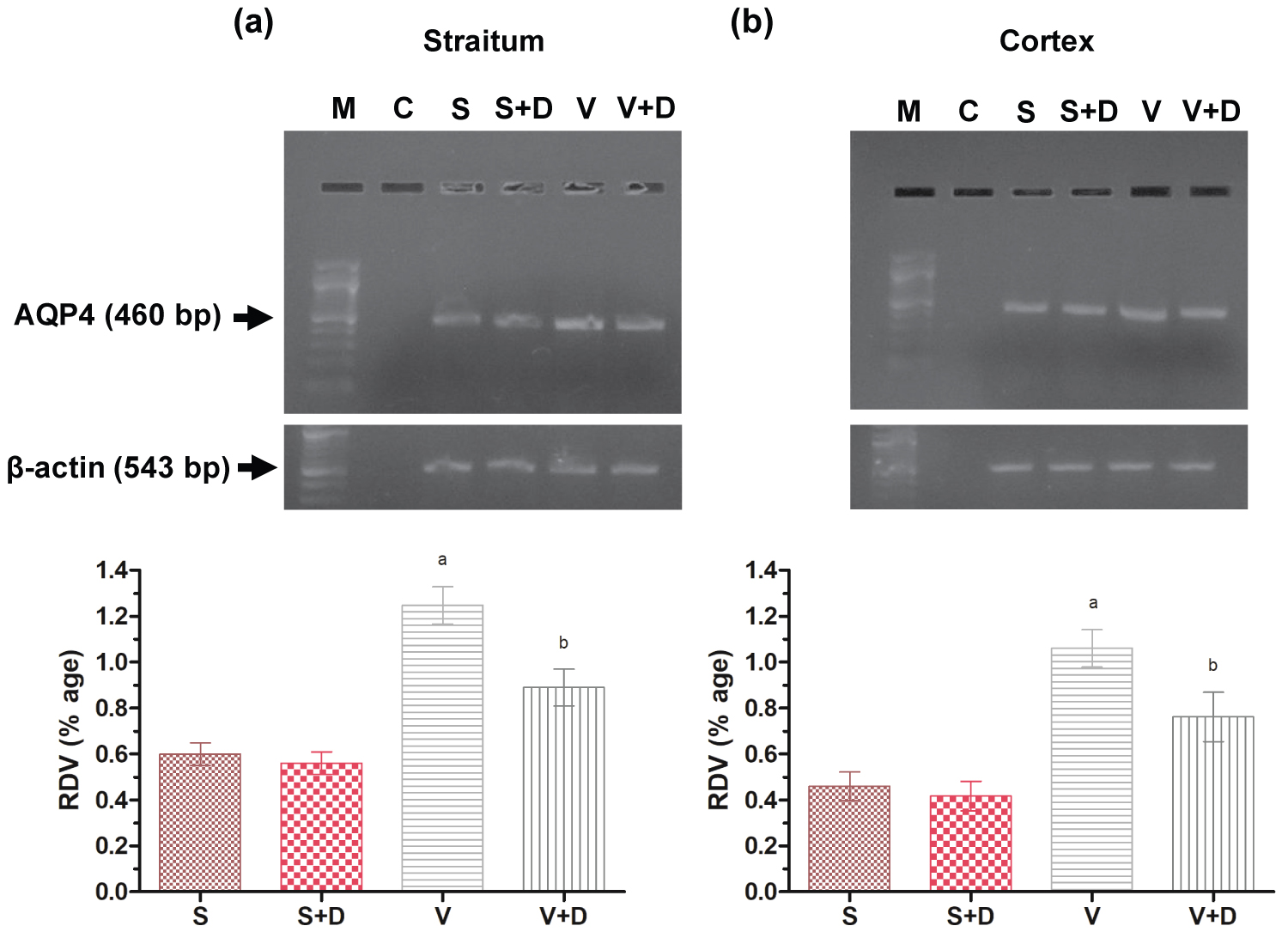
Semi-quantitative RT-PCR of AQP4
For amplification in linear range for semi-quantitative RT-PCR, the number of cycles considered for AQP4 was 29 and for β-actin, it was considered 26 as previously reported by Gupta, et al. [25]. Semi-quantitative RT-PCR of AQP4 of the cortical and striatal region (Figure 7a and Figure 7b) shows that its expression is significantly highest in vehicle (P ≤ 0.05) as compared to that of sham, and its expression is significantly low in vehicle with drug as compared to that of vehicle (P ≤ 0.05) [30-32].
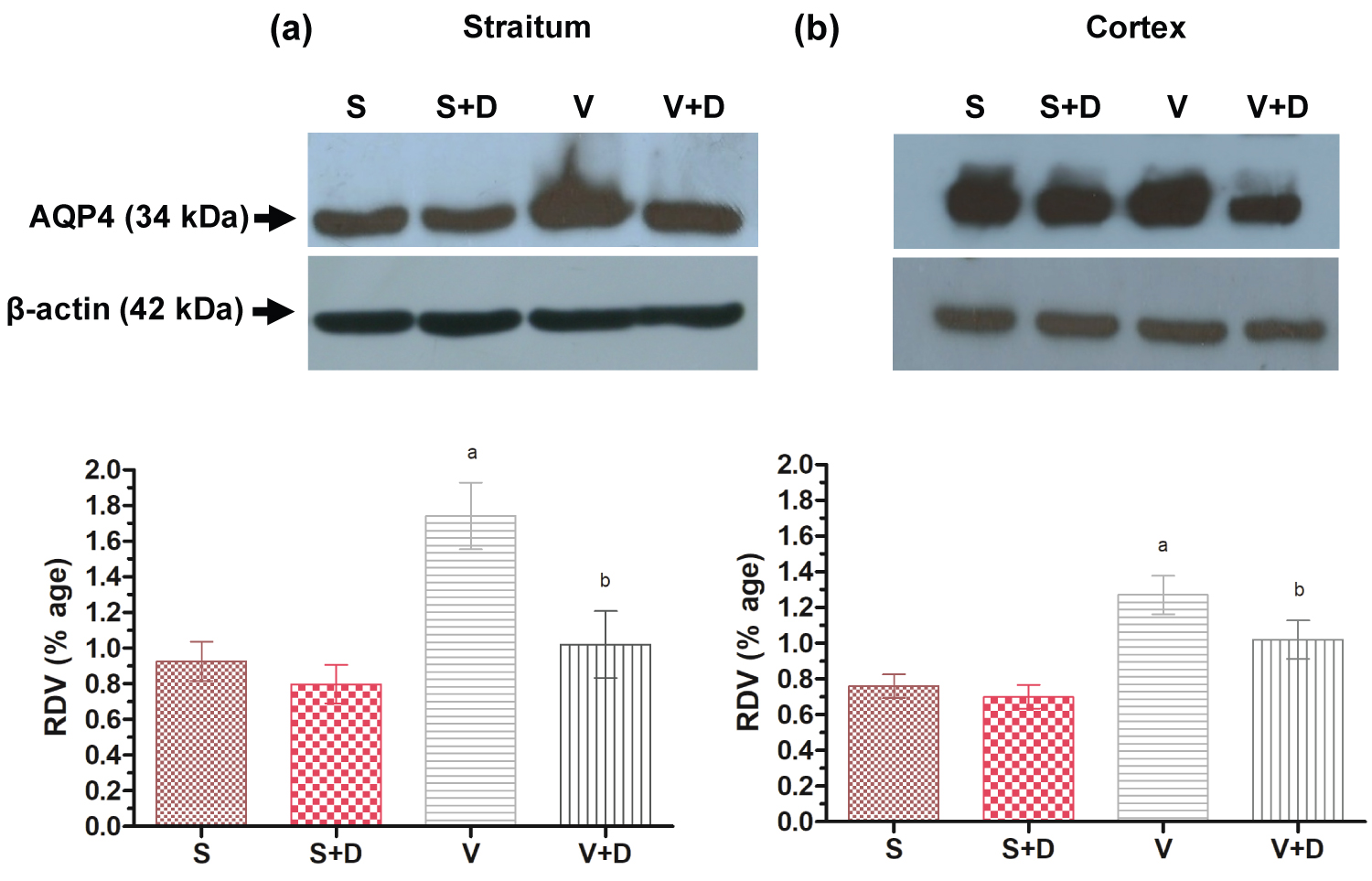
Western blot analysis of AQP4
For expression of aquaporin-4 protein, a rabbit polyclonal antibody specific to AQP-4 was used [30,31]. AQP-4 expression in the vehicle was found to be significantly (P ≤ 0.05) higher than the sham in both striatal and cortical region of the brain (Figure 8a and Figure 8b). Piroxicam supplemented vehicle cortex and striatum showed significant decrease in AQP4 level and proves the AQP4 attenuating property of Piroxicam.
Discussion
Ischemic insult culminates to a sequel of biochemical events like overload of calcium in neurons, NMDA receptor activation, inflammation, oxidative stress and apoptosis. Owing to increased calcium, enzymes like neuronal nitric oxide synthase (nNOS), phospholipase, calpain, ligase, xanthine oxidase and DNAses that are calcium dependent and involved in free radical production and catabolism of phoshpholipids, proteins and nucleic acids are activated. Further, reperfusion leads to generation of free radicals. These events lead to membrane breakdown leading to severe neuronal insult [33-35].
Pharmacological leads for stroke have been explored much but none of them could be translated as an efficient therapy. As suggested by the Stroke Therapy Academic Industrial Roundtable (STAIR), therapeutic agents with mixed pharmacology or sequential staging by combining different mechanistic approaches, might prove beneficial for stroke therapeutics [36].
Cyclooxygenase-2 (COX-2) isoforms were rampantly studied in ischemic stroke and COX-2 inhibitor administration have produced significant outcome in context to infarct volume reduction [37,38]. NSAID's, due to their property to inhibit prostaglandin E2 via cycloxygenase (COX) mediated prostaglandins synthesis may play a major role in addressing nociception and inflammation [39-42]. Although, COX independent actions of NSAIDs are also reported, such as flurbiprofen R12 enantiomer doesn't inhibit COX, however, they possess potent anti-inflammatory and antinociceptive effects which were later found to be mediated by inhibition of NFkB and AP-1 activation [43].
We selected Piroxicam, a NSAID, as a molecule of choice to explore its neuroprotective effects. Piroxicam as a neuroprotectant has already been established in vitro besides its antioxidant and anti-inflammatory property, which is a common characteristics of all NSAIDs [40,41]. Studies by Nakagomi, et al. reported piroxicam and flurbiprofen, at higher doses, ameliorated delayed neuronal death in the hippocampal CA sector after forebrain ischemia and affected hyperlocomotion [44]. However, no study has been undertaken to explore its role in modulating AQP4 water channel and reducing AQP4 mediated brain edema.
Our studies involved neuroprotective role of Piroxicam and its downstream effect on AQP4 expression in cortical and striatal regions of stroke brain. These regions were selected based on reports stating that following MCA occlusion, cells in the striatum die immediately [44,45]. Piroxicam administration should slow down or prevent the cell death in these areas. Piroxicam at a dose of 10 mg/kg b.w offered neuroprotection as demonstrated by the decrease in functional deficits, volume of infarct and improvement in motor function. Therefore, it seems as if Piroxicam is modulating the mechanism of neuronal damage, thus helping to decrease neuronal infarction progression.
Following a surge in neuronal calcium, protein kinase C regulated and calcium-calmodulin dependent kinase neuronal nitric oxide synthase (nNOS) gets activated. nNOS being a physiological mediator of vasodilation generates nitric oxide (NO) by increasing cGMP in smooth muscles. Following ischemia NO combines with superoxide anion to form peroxynitrite anion and causes neuronal insult. This anion further breakdowns into toxic nitrogen dioxide and hydroxyl radicals, exacerbating the neuronal injury [46]. In our study, a rise in nitrite levels in the insulted brain region was observed after 10 minutes following ischemic stroke and reduced to basal levels after 60 min [47,48]. A reduction in NO levels following Piroxicam administration could be associated with a reduction in calcium influx resulting from inhibition of ASIC1a as reported previously by us on the basis of in-silico studies [17,18], aiding towards neuroprotection.
The extent of lipid composed neuronal membrane damage can be determined by MDA as a biomarker. The peroxidation of lipid cascade in brain post ischemia occurs as a combined action of free radicals and NO generated following the activation of phospholipase A2 and nNOS [49]. An increase in MDA levels were observed immediately post reperfusion in the brain and is in accordance to previously reported studies [50]. Piroxicam mediated decrease in MDA suggests a possibility of attenuation of calcium influx involved in activation of phospholipase A2 and nNOS.
Cognitive, behavioral, hormonal and functional abnormalities are often associated with ischemic stroke [51]. Within the CA1 hippocampal regions, death of pyramidal neurons are responsible for the decline in cognitive functions such as memory and spatial learning following ischemic insult. Although, ischemia triggers proliferation of cells within the sub-granular zone, however newly formed neurons have a limited role to play in restoring behavioral impairments induced by ischemia [52-55]. Although, we have no substantial evidence to validate that Piroxicam plays a role in cognition, however we found that Piroxicam brought benefits in functional outcome and cognition, might be by targeting cellular mechanics [17,18]. Results from Rotarod experiment demonstrated that following ischemia, rats exhibited motor function impairments and lack of coordination. Rats with ischemic stroke showed a lower latency time to fall off time from the rotating rod as compared to control. Piroxicam treated stroke rats showed improvement in motor performance in rotarod test as compared to vehicle group. Findings from our study indicate that Piroxicam helps in lowering the time for spatial recognition.
Brain edema extends over days, hence interventions are required to reduce it. The role of water channels in endothelial cells causing edema is still unclear. Movement of water paracellularly after blood-brain barrier is disruption may take place because of the loss of the tight junctions as well [56,57]. Verkman and colleagues using aquaporin-4 knockout mice propounded the idea that aquaporin-4 channels might be an integral player towards cerebral edema after ischemic stroke. Experiments with AQP4-null mice suggested that AQP4 provides a pathway for water permeability in the brain [58,59]. Genetic knockout mice showed altered localization and expression of aquaporin-4 channels. AQP4 blocker has been a difficult task and none of the NSAIDs were able to inhibit water permeability through AQP4 [23,57]. We hypothesized that Piroxicam mediated AQP4 down regulation might decrease the formation of brain edema after ischemic stroke. Here we report chronic changes taking place in the expression of AQP4 in focal cerebral ischemia by Piroxicam that might affect formation of edema and subsequent tissue damage involved in defining overall functional deficits following ischemic stroke.
Previously we have performed molecular docking of AQP4 with water, its natural substrate and further with Piroxicam. The binding energy of Piroxicam was found to be significantly higher to its natural substrate in comparison [17,18]. Further, as per previous reported studies indicating a stable and maximum AQP4 expression 24 hr post ischemia, we performed molecular studies 1/24 hr reperfusion injury [28,29]. For checking AQP4 expression, cortex and straitum regions of the brain were considered. We also considered a study group as Sham + Drug to observe the after effect of Piroxicam in the expression of AQP4 in sham rats. Hence 4 study groups 1) Sham 2) Sham + Drug 3) Vehicle 4) Vehicle + Drug were considered. A significant increase in AQP4 expression was observed in the vehicle group as compared to both sham and Sham + Drug groups (Figure 7). If we look into the comparative outcome of Sham and Sham + Drug we find that Piroxicam does not affect the expression of AQP4 in normal pathology significantly. When the group administered with vehicle and Piroxicam is seen, AQP4 expression is decreased significantly (Figure 8). Our result clearly indicates that in focal cerebral ischemia, Piroxicam downregulates AQP4 expression which also justifies the earlier results obtained from molecular docking studies. Further, to explore the comparative AQP4 inhibition property by other NSAID like flurbiprofen whose neuroprotective role is well reported in past [26], we performed a comparative immunoblot (data not given). We found that the AQP4 inhibitory property of Piroxicam is optimal in comparison to flurbiprofen. Hence, we conclude that Piroxicam acts on AQP4 mediated brain edema in more effective way than other NSAID.
Mixed reports have been published regarding both upregulation and down regulation of AQP4 following ischemic stroke [60]. The variations in results may arise due to specie differences (rats versus mouse), animal age (adult versus young) or the degree of severity of ischemia/reperfusion (time variation in ischemia/reperfusion). AQP4 down-regulation in Piroxicam treated group was observed in our study, which was accompanied by a decrease in cerebral edema. Studies have reported that AQP4 over-expression increases water influx and precipitates the development of cytotoxic edema after ischemic stroke, hence, cerebral edema may be alleviated via AQP4 downregulation [5,25,61-63].
AQP4 phosphorylates protein kinase C and its activity gets altered with its phosphorylation [64]. NSAIDs are typically activators of protein kinase C which attenuate brain swelling induced following ischemia by inhibiting up-regulation of AQP4 [65]. Thus, it may be suggested that protein kinase C may be the target of Piroxicam, which during ischemia-reperfusion injury regulates AQP4 expression. Glutamate may also be one of the other factors as studies have shown that glutamate is responsible for swelling of astrocytes [66]. Alterations in the levels of brain glutamate were observed in AQP4 knockout mice. Nevertheless, NSAID by various mechanisms could decrease glutamate levels [59]. Therefore, Piroxicam via glutamate may also possibly regulate AQP4 expression. Significant increase in AQP4 expression after stroke may be related to AQP4 translocation towards the endfeet with no change in the overall abundance of AQP4. Thus, in the early phase AQP4 upregulation might be an intrinsic protective mechanism of the brain to facilitate more water movement. But at later point of time a limit to brain swelling is also required, thus AQP4 down regulation may bring the neuroprotective effect [67,68]. Hence we can infer from our results that AQP4 expression is effectively inhibited by Piroxicam, which can suppress brain edema formation and subsequently confer neuroprotection in a rodent model of focal cerebral ischemia.
Findings from the current study provide significant proof that Piroxicam acts as potent AQP4 regulator. Additionally, neuroprotective action of Piroxicam post ischemia enhances its clinical and curative potential. Mechanisms such as inhibition of NF-kB and COX activity may also in part contribute towards neuroprotection as observed in the case of other NSAIDs. Further, we will explore towards resolving of brain edema and maintain the normal equilibrium of AQP4 within the brain rather than to either stimulate or to block their action for good therapeutic strategies. In addition, it would be interesting to further evaluate its pharmacological proprieties and toxicological effects in large animals and humans.
Funding
IIT-BHU Departmental Funding.
Authors Contribution
PB, RP conceived the idea and design of the study. PB, AP, SP conducted the experiments. PB, AP, SP wrote the manuscript. RP finalized the manuscript.
Acknowledgments
The authors thank Padma Shri (Late) Prof. M.S. Kanungo and Mr. R.K Gupta of the Department of Zoology, BHU, India for their impartial support and inputs in the design of the work. Authors also thank Dr. Dileep R Yavagal, Deptt. Of Neurology, University of Miami Miller School of Medicine for the support and inputs on this study.
Conflict of Interest
The authors have declared that no competing interests exist.
Ethics Approval and Consent to Participate
NA.
Human and Animal Rights
Approval for animal use was taken from Institutional Animal Ethical Committee (Reg No.- 542/AB/CPCSEA;D/09-10/765).
Consent for Publication
All authors have given their consent for the publication of the manuscript.
References
- Manley GT, Binder D, Papadopoulos M, et al. (2004) New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience 129: 981-991.
- Papadopoulos MC, Krishna S, Verkman A (2002) Aquaporin water channels and brain edema. Mt Sinai J Med 69: 242-248.
- Tang G, Yang GY (2016) Aquaporin-4: A potential therapeutic target for cerebral edema. Int J Mol Sci 17: 1413.
- Nakano T, Nishigami C, Irie K, et al. (2018) Goreisan prevents brain edema after cerebral ischemic stroke by inhibiting aquaporin 4 upregulation in mice. Journal of Stroke and Cerebrovascular Diseases 27: 758-763.
- Amiry-Moghaddam M, Otsuka T, Hurn PD, et al. (2003) An a-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A 100: 2106-2111.
- Rash JE, Yasumura T, Hudson CS, et al. (1998) Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci U S A 95: 11981-11986.
- Manley GT, Fujimura M, Ma T, et al. (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6: 159-163.
- Chen JQ, Zhang CC, Jiang SN, et al. (2016) Effects of aquaporin 4 knockdown on brain edema of the uninjured side after traumatic brain injury in rats. Med Sci Monit 22: 4809-4819.
- Sun MS, Jin H, Sun X, et al. (2018) Free radical damage in ischemia-reperfusion injury: An obstacle in acute ischemic stroke after revascularization therapy. Oxid Med Cell Longev 2018: 2018.
- Wu L, Xiong X, Wu X, et al. (2020) Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front Mol Neurosci 13: 28.
- Chen Hs, Chen X, Li Wt, et al. (2018) Targeting RNS/caveolin-1/MMP signaling cascades to protect against cerebral ischemia-reperfusion injuries: Potential application for drug discovery. Acta Pharmacol Sin 39: 669-682.
- Xie W, Zhou P, Sun Y, et al. (2018) Protective effects and target network analysis of ginsenoside Rg1 in cerebral ischemia and reperfusion injury: A comprehensive overview of experimental studies. Cells 7: 270.
- Chance B, Sies H, Boveris A (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59: 527-605.
- Halliwell B (1987) Oxidants and human disease: Some new concepts. FASEB J 1: 358-364.
- Bhattacharya P, Pandey AK, Shukla SC, et al. (2013) Neuroprotection by µ-calpain and matrix metalloproteinases inhibition by Piroxicam in cerebral ischemia: An in silico study. Medicinal Chemistry Research 22: 5112-5119.
- Dorofeeva NA, Barygin OI, Staruschenko A, et al. (2008) Mechanisms of non-steroid anti-inflammatory drugs action on ASICs expressed in hippocampal interneurons. Journal of neurochemistry 106: 429-441.
- Bhattacharya P, Pandey AK, Paul S, et al. (2012) Cognitive effects of NSAIDs in cerebral ischemia: A hypothesis exploring mechanical action mediated pharmacotherapy. Med Hypotheses 79: 393-395.
- Bhattacharya P, Pandey AK, Paul S, et al. (2012) Neuroprotective potential of Piroxicam in cerebral ischemia: An in silico evaluation of the hypothesis to explore its therapeutic efficacy by inhibition of aquaporin-4 and acid sensing ion channel1a. Med Hypotheses 79: 352-357.
- Vartiainen N, Huang CY, Salminen A, et al. (2001) Piroxicam and NS-398 rescue neurones from hypoxia/reoxygenation damage by a mechanism independent of cyclo-oxygenase inhibition. J Neurochem 76: 480-489.
- Longa EZ, Weinstein PR, Carlson S, et al. (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20: 84-91.
- Dunham NW, Miya TS (1957) A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc. 46: 208-209.
- JK Callaway, MJ Knight, DJ Watkins, et al. (1999) Delayed treatment with AM-36, a novel neuroprotective agent, reduces neuronal damage after endothelin-1-induced middle cerebral artery occlusion in conscious rats. Stroke 30: 2704-2712.
- Pandey AK, Hazari PP, Patnaik R, et al. (2011) The role of ASIC1a in neuroprotection elicited by quercetin in focal cerebral ischemia. Brain Res 1383: 289-299.
- Hara H, Huang PL, Panahian N, et al. (1996) Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab 16: 605-611.
- Guevara I, Iwanejko J, Dembinska-Kiec A, et al. (1998) Determination of nitrite/nitrate in human biological material by the simple Griess reaction. Clin Chim Acta 274: 177-188.
- Chen W, Liu Jl, Sang Hf, et al. (2008) Therapeutic time window of flurbiprofen axetil's neuroprotective effect in a rat model of transient focal cerebral ischemia. Chin Med J (Engl) 121: 2572-2577.
- Nelson SK, Bose SK, McCord JM (1994) The toxicity of high-dose superoxide dismutase suggests that superoxide can both initiate and terminate lipid peroxidation in the reperfused heart. Free Radic Biol Med 16: 195-200.
- Pokorski M, Strosznajder R (1993) PO 2-Dependence of phospholipasec in the cat carotid body. Neurobiology and cell physiology of chemoreception. Springer 191-5.
- Frydenlund DS, Bhardwaj A, Otsuka T, et al. ( 2006) Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc Natl Acad Sci U S A 103: 13532-13536.
- Modi PK, Kanungo M (2010) Age-dependent expression of S100ß in the brain of mice. Cell Mol Neurobiol 30: 709-716.
- Gupta RK, Kanungo M (2013) Glial molecular alterations with mouse brain development and aging: Up-regulation of the Kir4. 1 and aquaporin-4. Age 35: 59-67.
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
- Chan PH (1996) Role of oxidants in ischemic brain damage. Stroke 27: 1124-1129.
- Fisher M, Hanley DF, Howard G, et al. (2007) Recommendations from the STAIR V meeting on acute stroke trials, technology and outcomes. Stroke 38: 245-248.
- Krueger M, Mages B, Hobusch C, et al. (2019) Endothelial edema precedes blood-brain barrier breakdown in early time points after experimental focal cerebral ischemia. Acta Neuropathologica Communications 7: 1-17.
- Kelso ML, Scheff NN, Scheff SW, et al. (2011) Melatonin and minocycline for combinatorial therapy to improve functional and histopathological deficits following traumatic brain injury. Neurosci Lett 488: 60-64.
- Huang H, Al-Shabrawey M, Wang MH (2016) Cyclooxygenase-and cytochrome P450-derived eicosanoids in stroke. Prostaglandins & other lipid mediators 122: 45-53.
- Liu S, Dai Ye, Zhou C, et al. (2020) Parecoxib exhibits anti-inflammatory and neuroprotective effects in a rat model of transient global cerebral ischemia. J Toxicol Environ Health A 83: 203-214.
- Chu XP, Miesch J, Johnson M, et al. (2002) Proton-gated channels in PC12 cells. J Neurophysiol 87: 2555-2561.
- Burgos-Ramos E, Puebla-Jiménez L, Arilla-Ferreiro E (2008) Minocycline provides protection against ß-amyloid (25-35)-induced alterations of the somatostatin signaling pathway in the rat temporal cortex. Neuroscience 154: 1458-1466.
- Ekdahl CT, Claasen JH, Bonde S, et al. (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A100: 13632-13637.
- Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231: 232-235.
- Tegeder I, Niederberger E, Israr E, et al. (2001) Inhibition of NF-?B and AP-1 activation by R-and S-flurbiprofen. FASEB J 15: 595-597.
- Nakagomi T, Sasaki T, Kirino T, et al. (1989) Effect of cyclooxygenase and lipoxygenase inhibitors on delayed neuronal death in the gerbil hippocampus. Stroke. 20: 925-929.
- Desai ID (1984) Vitamin E analysis methods for animal tissues. Methods Enzymol 105: 138-147.
- Suzuki J, Katoh N (1990) A simple and cheap methods for measuring serum vitamin A in cattle using only a spectrophotometer. Nihon Juigaku Zasshi 52: 1281-1283.
- Jagota S, Dani H (1982) A new colorimetric technique for the estimation of vitamin C using Folin phenol reagent. Anal Biochem 127: 178-182.
- Stirling DP, Koochesfahani KM, Steeves JD, et al. (2005) Minocycline as a neuroprotective agent. Neuroscientist 11: 308-322.
- Beckman JS, Beckman TW, Chen J, et al. (1990) Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 87: 1620-1624.
- Popp A, Jaenisch N, Witte OW, et al. (2009) Identification of ischemic regions in a rat model of stroke. PloS One 4: e4764.
- Al-Qazzaz NK, Ali SH, Ahmad SA, et al. (2014) Cognitive impairment and memory dysfunction after a stroke diagnosis: A post-stroke memory assessment. Neuropsychiatr Dis Treat 10: 1677-1691.
- Cendelín J, Korelusová I, Vožeh F (2008) The effect of repeated rotarod training on motor skills and spatial learning ability in Lurcher mutant mice. Behav Brain Res 189: 65-74.
- Bendel O, Prunell G, Stenqvist A, et al. (2005) Experimental subarachnoid hemorrhage induces changes in the levels of hippocampal NMDA receptor subunit mRNA. Brain Res Mol Brain Res137: 119-125.
- Kirino T (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239: 57-69.
- Pulsinelli WA, Brierley JB, Plum F (1982) Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 11: 491-498.
- Zheng YY, Lan YP, Tang HF, et al. (2008) Propofol pretreatment attenuates aquaporin-4 over-expression and alleviates cerebral edema after transient focal brain ischemia reperfusion in rats. Anesth Analg 107: 2009-2016.
- Migliati E (2006) Role of aquaporin-4 water channels in cerebral edema after ischemic stroke.
- Verkman A, Binder DK, Bloch O, et al. (2006) Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim Biophys Acta 1758: 1085-1093.
- Ma B, Xiang Y, Mu SM, et al. (2004) Effects of acetazolamide and anordiol on osmotic water permeability in AQP1-cRNA injected Xenopus oocyte. Acta Pharmacol Sin 25: 90-97.
- Hsu Y, Tran M, Linninger AA (2015) Dynamic regulation of aquaporin-4 water channels in neurological disorders. Croat Med J 56: 401-421.
- Katsuta K, Umemura K, Ueyama N, et al. (2003) Pharmacological evidence for a correlation between hippocampal CA1 cell damage and hyperlocomotion following global cerebral ischemia in gerbils. Eur J Pharmacol 467: 103-109.
- Galvao RI, Diógenes JP, Maia GC, et al. (2005) Tenoxicam exerts a neuroprotective action after cerebral ischemia in rats. Neurochem Res 30: 39-46.
- de Castro Ribeiro M, Hirt L, Bogousslavsky J, et al. (2006) Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res 83: 1231-1240.
- Lennon AM, Ramauge M, Pierre M (2002) Role of redox status on the activation of mitogen-activated protein kinase cascades by NSAIDs. Biochem pharmacol 63: 163-170.
- Casper D, Yaparpalvi U, Rempel N, et al. (2000) Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci lett 289: 201-204.
- Han BC, Koh SB, Lee EY (2004) Regional difference of glutamate-induced swelling in cultured rat brain astrocytes. Life Sci 76: 573-583.
- Fan Y, Zhang J, Sun XL, et al. (2005) Sex-and region-specific alterations of basal amino acid and monoamine metabolism in the brain of aquaporin-4 knockout mice. J Neurosci Res 82: 458-464.
- Gunnarson E, Zelenina M, Aperia A (2004) Regulation of brain aquaporins. Neuroscience 129: 945-953.
- Bhattacharya P, Pandey AK, Paul S, et al. (2013) Aquaporin-4 inhibition mediates piroxicam-induced neuroprotection against focal cerebral ischemia/reperfusion injury in rodents. PLoS One 8: e73481.
Corresponding Author
Ranjana Patnaik, School of Biomedical Engineering, Indian Institute of Technology, Banaras Hindu University, Varanasi- 221005 (U.P), India;
Pallab Bhattacharya, School of Biomedical Engineering, Indian Institute of Technology, Banaras Hindu University, Varanasi-221005 (U.P), India
Copyright
© 2020 Bhattacharya P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.