Cerebrotendinous Xanthomatosis: Literally an Orphan Disease in Brazil: Case Report
Abstract
Cerebrotendinous Xanthomatosis (CX) is an exceedingly rare autosomal recessive lipid storage disorder due to homozygous mutation of the mitochondrial enzyme sterol 27-hydroxylase, which is responsible for conversion of cholesterol to cholic and Chenodeoxycholic Acid (CheA), thus playing a key role in maintaining normal cholesterol levels in the body. Its clinical signs include juvenile cataracts, tendon xanthomas, premature atherosclerosis, and neurologic disturbances (dementia, pyramidal and extrapyramidal signs, ataxia, seizures, psychiatric disorders, and peripheral neuropathy). The disorder also may be associated with osteoarthritis, skeletal fracture, pes cavus, pulmonary insufficiency, endocrinopathy, renal and hepatic calculi, and childhood chronic diarrhea. Although it is extremely uncommon, it is easy to be diagnosed and its treatment with CheA, if instituted promptly, can avoid the progression of the disease. The present article is a report of a young patient, with a long evolution until the final diagnosis and it also reveals the difficulties of the proper treatment in countries like Brazil, where CheA is unavailable.
Keywords
Cerebrotendinous Xanthomatosis, Chenodeoxycholic acid, Neurology, Drug therapy
Introduction
Cerebrotendinous Xanthomatosis (CX) was described at the first time in 1937 by Van Bogaert, et al. [1] and is due an autosomal recessive inborn error which results in a mutation of the mitochondrial cytochrome P450 sterol 27-hydroxylase enzyme, which is responsible for the conversion of cholesterol to cholic and chenodeoxycholic acid, thus playing a key role in maintaining normal cholesterol levels in the body [2].
The lack of cholic and Chenodesoxycholic Acid (CheA) leads to absence of the negative feedback on the rate-limiting enzyme in bile acid synthesis (7 alpha-hydroxylase), and by way of a side pathway, occurs an accumulation of bile acid precursors, which are subsequently converted to cholestanol and bile alcohols, defining the laboratory hallmarks of CX: Increased serum cholestanol concentration and elevated urinary bile alcohols [3,4]. Besides that, patients with CX typically present normal or increased total cholesterol levels, decreased high-density lipoprotein cholesterol concentrations, increased 7α-hydroxy-4-cholesten-3-one (7Αc4), increased lathosterol, increased plant sterols (campesterol, sitosterol), increased serum and urinary bile acid alcohols but reduced serum CheA and 27-hydroxycholesterol (27-OHC) [5,6]. Because cholestanol is exclusively synthesized in the liver and not synthesized in the nervous system, another peculiarity of CX is the presence of cholestanol in the brain and cerebrospinal fluid. The exact mechanism is still unknown, but the presence of apolipoprotein B, which may carry cholestanol as well as cholesterol, in cerebrospinal fluid, indicates penetration of low-density lipoprotein through the blood-brain barrier [7].
In spite of being apparently easy to make the correct diagnosis, as an orphan disease, CX is frequently forgotten and in countries where laboratory support to define this disease is not fully available, it could be an enormous difficult task. Other major problem is that, sometimes, even the treatment was not available. Both situations could be quite challenging to physician and patients, as we will report.
Case Presentation
The data presented here were collected after the patient previous authorization and he also signed a consent term which allowed us to reveal his clinical data and images.
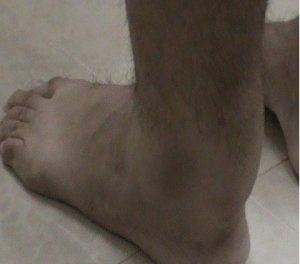
The patient was a man, twenty-four-year-old, white, and presumed parent consanguinity was suspected, but a well-defined family pedigree was unobtainable. A prior history of chronic diarrhea during his childhood, from birth until five years of age, but sporadically until the age of 15, was reported, with a presumed diagnostic of "lactose intolerance". A surgical treatment of "bilateral congenital cataracts" was also reported at the age of 8 years and a past history of learning difficulties, but without a well-defined mental impairment, as he graduated at high school regularly. Since childhood, a cavus foot was observed by his mother and also an unsteadiness gait. At the age of 10 years, a weakness was observed. Some simple tasks like turn faucet were troublesome. Orthopedic problems were started to be treated in his teenage and a "hypertrophic tendinitis" of the calcaneal tendons was noticed (Figure 1), confirmed by a biopsy performed by his orthopedic surgeon. Only at the age of 25, he was referred to a neurologist. In our first neurological evaluation, his neurological complaints were a gait disorder with a difficulty in negotiating curves, with paresthesia in both legs. The neurological examination also showed bilateral Babinski's sign with brisk patellar reflexes and ankle clonus. The ankle jerk reflexes were both abolished. Difficulties in high school learning process were also informed.
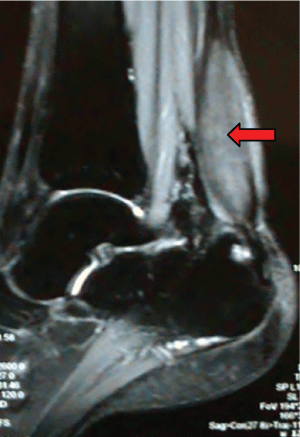
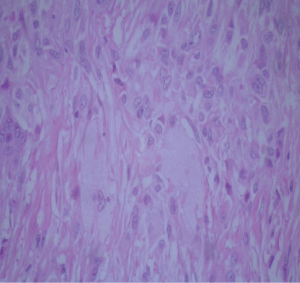
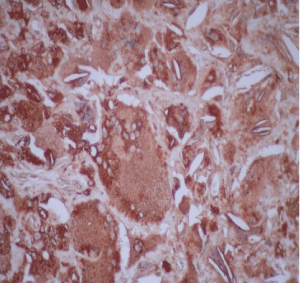
Diagnostic workup included an electroneuromyography, performed in EBNeuro Myto II 4 channel digital device with disposable monopolar needles. This exam confirmed a sensory and motor chronic axonal polyneuropathy. Brain, cervical and thoracic spine Magnetic Resonance Imaging (MRI) were initially unremarkable, but after a long period without proper treatment, 3 years after first exams, a spine imaging had been repeated and it had disclosed an extensive and longitudinal high signal abnormalities on T2 weighted MRI in posterior and lateral funiculi, addressing both gracile and cuneate fasciculi, and both corticospinal tracts. The ankles MRI revealed a hypertrophy of calcaneal tendons with presumed xanthomatous degeneration (Figure 2). Lately, the histology of the tendon confirmed an infiltration of xanthomatous histiocytes and the presence of cholesterol crystals interspersed to degenerate tendon (Figure 3 and Figure 4). A wide range general laboratory screening was unremarkable. Serum cholesterol was 178.1 mg/dl, High Density Lipoprotein (HDL) was 42.3 mg/dl; Very Low Density Lipoprotein (VLDL) was 23.6 mg/dl; low density lipoprotein was 112.3 mg/dl and triglycerides was 118 mg/dl.
Considering the high probability of a CX, determination of plasma cholestenol along with CYP27A1 gene study was conducted. No one of these tests was done in our country and the material was send to United States of America labs. The results have finally supported our diagnostic hypothesis, once high cholestenol level was observed (37.8 microg/ml for NR of 4.2 ± 1.2 microg/ml) and 7-dehydrocholesterol, 8-dehydrocholesterol and 8(9)-cholesterol were also slightly elevated. The CYP27A1 genetic study revealed homozygosis for mutation p.W268C:c.804G>T, compatible to CX. CheA therapy (dosage: 250 mg three times per day) was recommended, but only after a long term and huge judicial struggle, the patient has recently obtained the drug.
Discussion
The specifically mechanism how cholestanol accumulation involves the brain is still unknown. The presence of apolipoprotein B in cerebrospinal fluid explains this in part. Besides this, because of the diffuse decrease in total brain volume in most patients, it can be that the effects may be caused by increased apoptosis pathways [7].
As result of the accumulation of cholesterol and cholestanol in tissues the clinical manifestations of CX includes infantile-onset diarrhea (may be the earliest symptom), childhood-onset cataracts (can be the first finding in 75% of individuals), tendon xanthomas (specially of the Achilles tendon), progressive neurologic dysfunction (polyneuropathy, intellectual disability, psychiatric disturbance, pyramidal and/or cerebellar signs and seizures) and cardiovascular manifestations (like premature atherosclerosis) [2,3,7].
Early diagnosis is very important because the treatment can avoid disease progression and prevent permanent injury and new symptoms [2]. However, frequently the diagnosis is delayed with an average age at the time of diagnosis of thirty-five years [8]. CX must be investigated in all patients with tendon xanthomas in the presence of normal or slightly elevated cholesterol, with unexplained presence of bilateral juvenile cataracts and chronic diarrhea [7].
Clinically, the differential diagnosis is done with Marinesco-Sjogren syndrome, an autosomal recessive disorder characterized by cerebellar ataxia, congenital cataract and mental retardation, and with many other diseases like hypercholesterolemia, leukodystrophies, sitosterolemia, and Smith Lemli Opitz syndrome [3].
Replacement therapy with CheA is effective, safe, reduce clinical manifestations and plasma cholestanol levels [7]. This treatment suppresses synthesis of cholesterol, cholestanol, bile alcohols and bile acids [4]. The main adverse effects of this therapy are diarrhea [7]. Treatment with cholic acid is also effective for non-neurological symptoms [4]. If hypercholesterolemia is not controlled with CheA treatment alone, HMG-CoA reductase inhibitors (statins) may be added, although their effectiveness is controversial [3,7]. Surgical excision of xanthomas is not indicated because it may worsen the gait imbalance [4].
Molecular genetic analysis, accomplished in our patient, is obtained by the screening of CYP27A1 gene, a member of the cytochrome P450 gene family, located at the chromosome 2q33. The genomic structure of CYP27A1 contains nine exons and eight intones with 18.6 kb of DNA. The mature enzyme consists of four hundred ninety-eight amino acids and it is expressed in central nervous system, liver, lungs, duodenum and endothelial cells. Until now, more than fifty mutations were described in the CYP27A1 gene, usually affecting heme or adrenodoxin related domains between exons six and ninety-one [5,9]. The most common mutation changes the amino acid arginine to the amino acid cysteine at position three hundred sixty-two in the protein. The patients described here presented a homozygous CYP27A1 gene mutation in exon six (1183C>T) that was already described in other populations [9].
The clinical findings in the present patient were similar to other descriptions, but the main feature of our case report is that, in spite of being a very treatable condition, the difficulties about the CX treatment in our country are huge and have been discussed already in other articles [10,11]. As far as our knowledge can reach, there are only ten patients previously reported in Brazilian literature, and the main problem is the lack of CheA available in Brazil. Other major problem is also observed in our country: The diagnostic laboratory workup is not currently available and all the definitive and conclusive investigation must be done in other countries, with a possible time gap between the suspected clinical hypothesis and the laboratory supported diagnosis. The article wrote by Langue, et al. [10] also mentioned the difficult about the CYP27A1 analysis, which also was made in another country.
The present case had followed this "rule", once its entire laboratory workup was performed in United States of America, which undoubtedly had promoted a little time gap in defining the lab diagnosis. Besides, the patient had to pursue hardly the CheA treatment, a nonexistent drug in Brazil, and, to make things worse, our drug regulatory federal agency did not (and still does not) provide easy conditions to perform a regular and a fast drug importation. Only after a judicial injunction, which lasted more than one year, he had finally received the therapy, with unknown possible consequences to disease evolution.
Acknowledgements
Our gratefulness to Alexandre Galvão Bueno, M.D., for providing the photos of the tendon biopsy. To Prof Gerald Salem, M.D. of Division of Gastroenterology, New Jersey Medical School, who gently helped us with diagnostic resources in USA and who had also sent to the patient an initial course of CheA treatment. Thanks to the patient who allowed us to publish his clinical data.
References
- Van Bogaert L, Scherer HJ, Froehlich A, et al. (1937) Une deuxime observation de cholesterinose tendineuse symetrique avec symptoms cerebraux. Ann Med 42: 69-101.
- Di Taranto MD, Gelzo M, Giacobbe C, et al. (2016) Cerebrotendinous xanthomatosis, a metabolic disease with different neurological signs: Two case reports. Metab Brain Dis 31: 1185-1188.
- Saxena V, Pradhan P (2016) Cerebrotendinous xanthomatosis; A genetic condition: Clinical profile of three patients from a rural Indian family and review of literature. J Clin Orthop Trauma 7: 122-126.
- Patni N, Wilson DP (2016) Cerebrotendinous Xanthomatosis. Endotext 1-9.
- Lamon-Fava S, Schaefer EJ, Garuti R, et al. (2002) Two novel mutations in the sterol 27-hydroxylase gene causing cerebrotendinous xanthomatosis. Clin Genet 61: 185-191.
- Razi SM, Gupta AK, Gupta DC, et al. (2016) Cerebrotendinous xanthomatosis (a rare lipid storage disorder): A case report. Journal of Medical Case Reports 10: 1-5.
- Moghadasian MH, Salen G, Frohlich JJ, et al. (2002) Cerebrotendinous xanthomatosis: A rare disease with diverse manifestations. Arch Neurol 59: 527-529.
- Muniaswamy M, Rengasamy M, Aravamuthan R, et al. (2016) Cerebrotendinous xanthomatosis. Indian Dermatology Online Journal 7: 336-338.
- Verrips A, Hoesfloot LH, Steenbergen GCH, et al. (2000) Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain 123: 908-919.
- Lange MC, Zétola VF, Teive HAG, et al. (2004) Cerebrotendinous xanthomatosis: Report of two Brazilian brothers. Arq Neuro-Psiquiatr 62: 1085-1089.
- Cerqueira ACR, Nardi AE, Bezerra MF (2010) Cerebrotendinous xanthomatosis: A treatable hereditary neuro-metabolic disease. Clinics (Sao Paulo) 65: 1217-1218.
Corresponding Author
Emanuelle Bianchi da Silva, Department of Medicine, State University of Western Parana, Carlos Bartolomeu Cancelli Street, N 950, Cascavel City, Parana, Brazil, Zip Code: 85811-280, Tel: +55-045-99957-8504.
Copyright
© 2017 da Silva EB, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.