Therapeutic Agents Targeting at AGE-RAGE Axis for the Treatment of Diabetes and Cardiovascular Disease: A Review of Clinical Evidence
Abstract
Advanced Glycation End Products (AGEs), together with its receptor (RAGE) are known to play a predominant role in the onset of diabetic micro- and macrovascular diseases. Therefore, therapeutic interventions which can target AGE-RAGE axis are of great interest in diabetic therapy. Animal studies demonstrated promising results for most of the AGE-RAGE inhibitors but their actual clinical values are not fully elaborated. Therefore, this review aimed to summarize clinical findings of well-known AGE-RAGE antagonists including AGE cross-link breaker (alagebrium), dicarbonyl scavengers (aminoguanidine), antidiabetic drugs (metformin, thiazolidinediones, meglitinides, sulfonylureas and dipeptidyl peptidase 4 inhibitor), lipid-lowering drugs (statins), antihypertensive agents (angiotensin receptor blockers, angiotensin converting enzyme inhibitors and calcium channel blockers), vitamin B1 (thiamine and benfotiamine) and B6 (pyridoxine and pyridoxamine) as well as other investigational pharmacotherapy. The inhibitory mechanism of the anti-AGE-RAGE agents are also discussed. To date, most of the therapeutic interventions targeting at AGE-RAGE axis produced conflicting clinical findings. Some of the most promising agents are metformin, thiazolidinediones and statins. It is postulated that the inhibition of AGE-RAGE axis may be more beneficial at the early stage of diabetes so as to delay the progression of the associated vascular complications. Future studies should aim to identify the subsets of diabetic patients whom will be truly benefited from AGE-RAGE inhibition.
Keywords
Glycation, Maillard reaction, Nephropathy, Neuropathy, Reactive carbonyl species, Statin, Thiazolidinedione
Abbreviations
ACE: Angiotensin Converting Enzyme; ADAM10: A Disintegrin and Metalloproteinase 10; AGE: Advanced Glycation End Product; ARB: Angiotensin Receptor Blocker; CCB: Calcium Channel Blocker; CML: Carboxymethyllysine; DPP4: Dipeptidyl Peptidase 4; ECM: Extracellular Matrix; esRAGE: Endogenous Secretory Receptor for Advanced Glycation End Product; HbA1c: Glycated Haemoglobin A1c; ICAM-1: Intercellular Adhesion Molecule-1; MAPK: Mitogen-Activated Protein Kinase; NF-κB: Nuclear Factor-Kappa B; PI3K-Akt: Phosphatidylinositol-Protein Kinase B; PPAR: Peroxisome Proliferator-Activated Receptor; RAAS: Renin-Angiotensin-Aldosterone System; RAGE: Receptor for Advanced Glycation End Product; RCT: Randomized Controlled Trial; sRAGE: Soluble Receptor for Advanced Glycation End Product; T1DM: Type 1 Diabetes Mellitus; T2DM: Type 2 Diabetes Mellitus; TZD: Thiazolidinedione; VCAM-1: Vascular Cell Adhesion Molecule-1; VEGF: Vascular Endothelial Growth Factor
Introduction
Diabetes mellitus which is one of the most widespread chronic metabolic diseases is swiftly transforming into global health threat due to the ever-increasing prevalence of modern risks, namely high blood glucose, physical inactivity, overweight and obesity [1]. In 2014, it was estimated that about 422 million adults were diabetic, approximately 90% of which were Type 2 Diabetes Mellitus (T2DM) [2]. Diabetic vascular complications, notably the macrovascular diseases like atherosclerosis and microvascular diseases like retinopathy, nephropathy and neuropathy are the major contributors of morbidity and mortality in diabetes. In this context, Advanced Glycation End Products (AGE) and the interaction with the Receptor for AGE (RAGE) has been long recognized to be one of the key pathological pathways driving the progression of diabetes and the associated vascular complications.
AGEs are irreversibly generated from the non-enzymatic glycation of reducing sugars to amino groups in proteins or lipids, followed by a series of rearrangements or oxidation reactions [3]. This process, which is known as the Maillard reaction, is concentration-dependent since the circulating AGE level is significantly higher in diabetic patients [4]. Along with the elevation of AGE, many reactive intermediates such as glyoxal, methylglyoxal and 3-deoxyglucosone which are collectively known as α-oxoaldehydes or dicarbonyls, are also increased [5]. These dicarbonyl compounds can be derived from polyol pathway [6], degradation of protein glycation intermediates [7] or lipid peroxidation and oxidative stress [8]. Under high carbonyl stress, these dicarbonyls act as the AGE precursors and promote the formation and accumulation of AGEs [9].
The detrimental effects of AGEs have been summarized comprehensively in several reviews [10,11]. Two major mechanisms are implicated: (1) The formation of cross-linked products between AGEs in basement membrane of Extracellular Matrix (ECM); and (2) Ligand binding of AGEs to RAGE which triggers downstream cellular signaling. The former mechanism modifies the properties of structural proteins like collagen, vitronectin and laminin which subsequently, weakens the integrity of vascular structure and leads to vascular stiffening [12] and myocardial dysfunction [13]. The AGE-RAGE interaction, on the other hand, triggers the activation of various signaling cascades, resulting in functional changes in terms of proinflammatory response, programmed cell death, migration and proliferation [14]. AGE-mediated RAGE activation also triggers a positive feed forward loop, in which RAGE signaling activates NF-κB which further upregulates RAGE expression [15]. This self-perpetuating cycle intensifies the pathological progression of diabetic vascular diseases.
Considering the deleterious effects of AGEs, on-going investigations are being carried out to identify compounds that can inhibit the AGE-RAGE axis. Some of these AGE-RAGE inhibitors were tested in human clinical trials to examine their therapeutic effects on diabetes and diabetes-associated complications. Thus, the primary aim of this review was to outline the current clinical evidence of therapeutic drugs on AGE-RAGE axis intervention with a major focus on diabetes-related health conditions. The putative mechanisms of action of the experimental drugs on the glycation pathway are also discussed.
Pathological Roles of AGE-RAGE Axis in Diabetic Vascular Complications
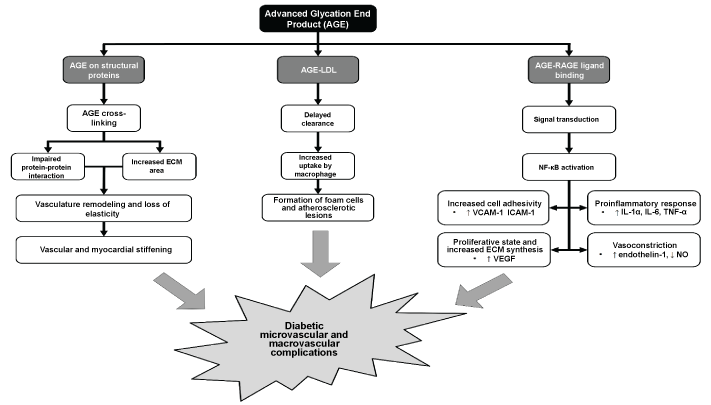
As mentioned previously, AGE and RAGE can induce both intra- and extracellular pathological changes which collectively, contribute to the development of endothelial damage, vasculature modification, proatherogenic and proinflammatory processes. Consequently, the cardiovascular function is tremendously jeopardized, resulting in vascular complications commonly seen in diabetic patients. The intra- and extracellular pathogenesis of AGE-RAGE axis is illustrated in Figure 1.
Generally, the extracellular effects of AGE accumulation are largely receptor-independent, but some of which can be aggravated by the activation of RAGE. Under physiological conditions, AGE accumulation is a normal aging process [16], but in diabetic patients, the process takes place at an accelerated pace. Non-enzymatic glycation can occur to virtually all proteins, lipids and nucleic acids, the last of which is less well-understood in diabetes mellitus. However, recent studies do suggest potential implication of glycated DNA in diabetic patients [17,18].
On the other hand, the impacts of protein glycation is extensively studied. AGE formation on the serum albumin can affect their drug binding capacity [19] and ability to induce platelet aggregation [20]. Furthermore, preliminary studies of glycated immunoglobulins revealed significant conformational alteration and loss of antibody-antigen interaction [21,22]. AGE aggregation is more prominent in structural proteins due to their slow turnover rate. These proteins, namely collagen, vitronectin and laminin, are responsible for the formation of basement membrane in the ECM. Increased AGE accumulation can modify the structural properties of the large matrix formed by these proteins via AGE-AGE interaction, or cross-linking [23]. These cross-links on the structural proteins increase the area of ECM [24] and hinder protein-protein interaction [25,26] which subsequently reduce their elasticity, leading to vascular and myocardial stiffening in diabetes and aging [27]. Moreover, AGE formation on Low-Density Lipoproteins (AGE-LDL) significantly impedes their clearance [28] and enhances their likelihood to be taken up by macrophages [29]. This promotes the formation of foam cells and atherosclerotic lesions, eliciting increased risk of atherosclerosis in diabetic patients.
Intracellularly, upon AGE-RAGE ligand binding, a series of signal transduction pathways are activated, among which are Mitogen-Activated Protein Kinase (MAPK), p21ras, Src kinase, JAK-STAT, protein kinase C and phosphatidylinositol-protein kinase B (PI3K-Akt) pathways [14]. These signaling cascades modify the cellular response to apoptosis, autophagy, proliferation, motility and inflammation. One of the key proinflammatory transcription factors, Nuclear Factor-Kappa B (NF-κB) is also activated. The activated transcription factor will translocate into the nucleus to upregulate the expression of various downstream target genes. In the endothelial cells, RAGE-induced NF-κB activation promotes cell-cell and cell-ECM adhesion via the overexpression of Intercellular Adhesion Molecule-1 (ICAM-1) and Vascular Cell Adhesion Molecule-1 (VCAM-1) respectively [30,31]. When coupled with NF-κB-dependent overexpression of Vascular Endothelial Growth Factor (VEGF) which facilitates ECM thickening, the increased vascular adhesiveness greatly enhances leucocyte retention, infiltration and activation [32]. RAGE activation also mediates proinflammatory response via the release of cytokines like interleukins and tumor necrosis factor α [33]. Such a RAGE- and NF-κB-induced proliferative and inflammatory state leads to substantial vascular remodeling and chronic vascular insult, both of which act as the key pathology to the onset of atherosclerosis and endothelial failure in diabetic microvascular complications [34-36].
RAGE activation also plays a role in vasoconstriction by stimulating the overexpression of a potent vasoconstrictor, endothelin-1 [37]. Furthermore, AGE aggregation has been shown to impair the biosynthesis of nitric oxide which is a crucial vasodilator. The inhibitory effect on nitric oxide bioavailability is caused by transcriptional suppression and direct inhibition of endothelial nitric oxide synthase [38,39]. The cumulative effect of reduced nitric oxide and elevated endothelin-1 impairs the blood perfusion considerably, resulting in a hypoxic state and progressive ischaemic injury to the peripheral tissues [40-42] (Figure 1).
Therapeutic Agents Targeting at AGE-RAGE Axis
AGE cross-link breaker and dicarbonyl scavenger
Some of the most extensively studied pharmacological interventions acting on AGE-RAGE axis are dicarbonyl scavengers (aminoguanidine) and AGE cross-link breaker (alagebrium). The clinical efficacy of these compounds are summarized in Table 1.
Aminoguanidine (or pimagedine) is an investigational drug that inhibits AGE formation by scavenging AGE precursors. It has two crucial functional groups, namely a nucleophilic hydrazine and a guanidino groups, both of which facilitate irreversible interaction with dicarbonyls, especially glyoxal, methyglyoxal and 3-deoxyglucosone [43]. Such a scavenging effect of aminoguanidine has been demonstrated to rescue diabetic nephropathy in diabetic animal models via the reduction of albuminuria and renal vascular injury [44,45].
Nonetheless, a randomized controlled clinical trial (ACTION I) which involved 690 patients with Type 1 Diabetes Mellitus (T1DM) failed to establish beneficial effects of aminoguanidine on diabetic nephropathy as there was no significant delay in serum creatinine doubling time between treated and untreated individuals [46]. Aminoguanidine regimen did however, slow down the deterioration of glomerular filtration rate and reduce total urinary proteinuria [46]. Another similar trial, ACTION II, which was designed to study the effects of aminoguanidine on diabetic renal complications among the patients with type 2 diabetes mellitus, was subjected to early termination due to safety issues and low efficacy of the drug [47].
The use of aminoguanidine is associated side effects like autoantibody generation, anemia, flu-like symptoms and very rarely, crescentic glomerulonephritis [46]. These could be linked to other biological functions of aminoguanidine. Essentially, aside from being a dicarbonyl scavenger, aminoguanidine is also a potent inhibitor of inducible nitric oxide synthase and diamine oxidase as well as a strong scavenger for multiple metabolites including pyridoxal phosphate, pyruvate and glucose [43,48]. Flu-like symptoms may be attributable to histamine intolerance caused by the inhibitory effect on diamine oxidase. On the other hand, since aminoguanidine is a hydrazine derivative, this may trigger the induction of autoimmunity which contributes to the onset of glomerulonephritis [49]. In light of these adverse effects, the clinical use of aminoguanidine may not be feasible. In fact, the development of aminoguanidine as a therapy for diabetic neuropathy has been halted due to the aforementioned toxicity.
As for the AGE cross-link breaker, alagebrium, Randomized Controlled Trials (RCTs) has yielded mixed results on its efficacy. Treatment with alagebrium (200-210 mg) could reduce arterial and left ventricular stiffness [50,51], but failed to confer beneficial effect on overall cardiovascular health [52-54]. Two smaller and single-arm studies showed that the therapeutic effects of alagebrium on cardiovascular function were more prominent at much higher dosage (420 mg) [55,56]. Based on these findings, treatment with alagebrium seems to provide certain mechanical improvements to the heart and major blood vessels. This is linked to its selective AGE cross-links cleavage properties, which attenuates AGE accumulation and reduces existing AGE cross-links in the vascular wall and myocardium [57]. As a result, the reduction of AGE cross-links helps to restore vascular and ventricular elasticity. Nevertheless, the overall cardiovascular fitness remained largely unaffected by alagebrium. It is not unusual given that diabetes- or ageing-related cardiovascular disease is a multifactorial disease. This also suggests that the clinical role of AGE cross-link breaker should be adjunctive rather than primary.
Unlike aminoguanidine, there is no serious adverse effect associated with alagebrium. Another AGE cross-link breaker, TRC4186 which has undergone phase I clinical study, was also reported to be safe and well-tolerated in human subjects [58]. Unfortunately, like aminoguanidine, the development of alagebrium has been discontinued after the company incharge stopped their operation. Likewise, there is no further update about TRC4186 despite the completion of phase II clinical trial in diabetic patients with stable heart failure (Table 1).
Anti-hyperglycaemic medications
Many classes of antidiabetic drugs such as biguanides, Thiazolidinediones (TZDs), meglitinides, sulfonylureas and Dipeptidyl Peptidase 4 (DPP4) inhibitor, have been tested clinically for their inhibitory effects on AGE-RAGE axis (Table 2). All of them have demonstrated a promising AGE-lowering effect. This is plausible as the reduction of glucose level favors the reverse reaction of glycation and hence, lowering the formation and aggregation of AGEs. However, it is postulated that some of these drugs may suppress the AGE-RAGE axis independent of their glucose-lowering properties.
Considering the structural similarity to aminoguanidine, metformin is also thought to serve as a dicarbonyl scavenger apart from its insulin-sensitizing effect. Indeed, metforrmin is capable of reducing methylglyoxal in vitro [59] and in T2DM patients [60]. The combined effect of the two bioactivities may contribute to the significant decline in circulating AGE level [61,62]. Nevertheless, compared to other glucose-lowering drugs like pioglitazone and repaglinide, metformin did not perform better in reducing AGEs [63,64]. This implies that the glycaemic control plays a more predominant role than dicarbonyl scavenging in AGE-RAGE axis inhibition. Aside from AGE-lowering effect, treatment with metformin also restored antioxidant capacity of the serum, reduced proinflammatory biomarkers and enhanced nitric oxide level [63,64]. Hence, metformin may be an attractive candidate for further study about AGE-RAGE inhibition and diabetic vascular complications.
Another anti-diabetic agent which has been rigorously tested is TZD. Fundamentally, TZDs like pioglitazone and rosiglitazone are Peroxisome Proliferator Activated Receptor (PPAR) agonist, with the highest preference for PPARγ isoform [65]. Studies revealed that the activation of PPARγ effectively down regulates the RAGE expression in the endothelial tissues [66,67]. This can further diminish NF-κB activation which helps to inhibit many downstream implications such as prothrombotic and proinflammatory responses of the blood vessels [3]. Two clinical trials reported significant increase in soluble RAGE (sRAGE) level with the treatment of rosiglitazone [68] and pioglitazone, in which the sRAGE-inducing effect of the latter was more prominent and rapid [69].
Principally, sRAGE originates from two sources, namely the proteolysis cleavage from membrane-bound RAGE [70] and the alternative splice variant of RAGE which is also known as endogenous secretory RAGE (esRAGE) [71]. Compared to the full-length RAGE isoform, sRAGE does not have the transmembrane and cytoplasmic signaling domains. As a result, ligand binding to sRAGE is unable to trigger signaling cascades. Such a distinctive feature makes sRAGE an effective decoy and competitive inhibitor of its membrane-bound counterpart which allows sRAGE to scavenge circulating AGEs, facilitate AGE detoxification and attenuate the pathological processes induced by RAGE activation [72]. Moreover, in T2DM patients, sRAGE was negatively correlated to RAGE expression, strongly indicative of a beneficial role of sRAGE in modulating AGE-RAGE axis [73]. Therefore, even though how exactly TZDs increase circulating sRAGE is largely unclear, such a bioactivity may mediate AGE disposal and suppress RAGE activation.
Despite the promising AGE-RAGE inhibitory effect of TZD, it is important to highlight that the sample size of the clinical trials was too small to draw a conclusive remark about its efficacy. Larger trials are therefore, warranted to fully elucidate the clinical prospect of TZDs in targeting AGE-RAGE axis. In addition to metformin and TZDs, the effects of repaglinide (meglitinide), glimepiride (sulfonylurea) and alogliptin (DPP4 inhibitor) on AGE-RAGE axis have also been examined [63,74,75]. Their AGE-lowering effect was encouraging and comparable to metformin and TZDs. However, the studies are also limited by their small sample sizes. In short, anti-hyperglycaemic drugs are potent AGE inhibitors in concordance to their glucose-lowering effects. Future studies are indispensable and should emphasize on their effectiveness on delaying the progression of diabetic micro- and macrovascular complications (Table 2).
Lipid-lowering drugs
Statins, notably atorvastatin, simvastatin, pravastatin and pitavastatin have been consistently found to inhibit AGE-RAGE axis. Briefly, HMG-CoA reductase inhibitor or so-called statin is one of the most commonly prescribed lipid-lowering medications for patients with hypercholesterolaemia due to its high efficiency to block conversion of HMG-CoA to mevalonic acid in cholesterol biosynthesis [76]. Recent trials (Table 3) ubiquitously reported a decline in circulating AGE level or elevation of sRAGE level following statin treatment [77-81]. AGE formation and RAGE expression in atherosclerotic plaques were also suppressed by simvastatin regimen [82].
The AGE-lowering effect by statins seems to be independent of glycaemic control because the glucose level and glycatedHaemoglobin (HbA¬1c) remained unchanged by statins in the studies outlined in Table 3. It is hypothesized that statins may induce RAGE shedding to facilitate increased sRAGE and AGE disposal [83]. Indeed, the results showed that statin stimulated proteolytic cleavage of membrane-bound RAGE by a disintegrin and metalloproteinase 10 (ADAM10) to yield sRAGE and this mechanism was proved to be strictly caused by cholesterol biosynthesis attenuation instead of isoprenylation inhibition [83]. Statin-induced RAGE shedding is not entirely a novel mechanism. Previous studies have pointed out that cellular cholesterol depletion can trigger ADAM10-facilitated shedding of interleukin-6 receptor [84], soluble amyloid precursor protein [85] and CD44 [86]. Proteolytic cleavage of these membrane-bound proteins correlates the roles of cellular cholesterol to inflammatory response, Alzheimer's disease prevention and suppression of tumor migration respectively. In this context, long term statin therapy may lead to cellular cholesterol depletion which in turn, promotes RAGE proteolytic shedding, although exact pathway remains uncertain. Consequently, circulating sRAGE level increases to scavenge AGEs and attenuate RAGE-mediated downstream signaling.
It is widely acknowledged that oxidative stress plays a critical role in the formation of advanced glycoxidation and lipoxidation end products [87]. In this context, statins also possesses antioxidant properties. It has been demonstrated that statin can inhibit superoxide generation from NADPH oxidase via the transcriptional suppression of NADPH oxidase subunits and blockade of NADPH oxidase activation [88-90]. Furthermore, treatment with statins could also alleviate AGE-induced cellular signaling pathways like NF-κB and MAPK which resulted in reduced reactive oxygen species production [91]. As a result, the reduced oxidative stress may inhibit AGE formation and RAGE-mediated proinflammatory signaling, which contribute to the beneficial effects observed in clinical studies.
As opposed to statins, the effect of fibrates (another class lipid-lowering drug) on AGE-RAGE axis is poorly understood. Thus far, no clinical evidence is available. In vitro and in vivo studies on this aspect is also limited. However, fibrates have been demonstrated to attenuate AGE-induced NF-κB activation in glomerular microvascular endothelial cells [92]. Such an inhibitory on NF-κB activation was also found in atherosclerosis-prone, diabetic mice, but whether or not the effect was AGE-dependent is unclear [93]. Nevertheless, two recent randomized controlled trials (ACCORD Eye study and FIELD study) unraveled the promising therapeutic effect of fibrates on diabetic retinopathy although the underlying mechanism remains unclear [94,95]. These exciting clinical findings justify future investigations of fibrates on AGE-RAGE axis.
As such, our knowledge about the effect of fibrates on the glycation pathway is lacking, but recent clinical studies may suggest possible therapeutic effects. Conversely, statins are well-studied and have a huge potential to be AGE-lowering agents. The RAGE shedding mechanism is also unique to HMG-CoA reductase inhibitors. Clinically, statins are known to associate with very few side effects. Hence, all these favorable features make statins a practical and excellent choice to be developed as a therapeutic intervention targeting AGE-RAGE axis in diabetic vasculopathy (Table 3).
Anti-hypertensive agents
Table 4 summarizes clinical studies on AGE-inhibiting effects of anti-hypertensive drugs of different pharmacological classes like Angiotensin Receptor Blockers (ARBs), Angiotensin Converting Enzyme (ACE) inhibitors and Calcium Channel Blockers (CCBs). Among the three classes, ARBs are the most extensively studied drugs on AGE-RAGE axis. Basically, angiotensin II receptors play an integral role in Renin-Angiotensin-Aldosterone System (RAAS), a hormone system that regulates blood pressure, electrolyte and water balance. Blocking the receptor results in vasodilation, reduction in aldosterone and catecholamine secretion as well as reduced water reabsorption which collectively lower blood pressure [98].
Regarding the inhibitory effect of ARBs on AGE-RAGE axis, clinical trials produced mixed results. Small and short (6 months) trials indicated that Carboxylmethyllysine (CML) and pentosidine were significantly reduced in hypertensive patients by valsartan and olmesartan [99,100]. This is further supported by another single-arm study which reported decreased serum AGEs after valsartan therapy in Japanese T2DM patients with hypertension [101]. In contrast, larger RCTs with longer follow-up duration (2 to 4 years) concluded that treatment with ARBs did not lower AGEs and other AGE adducts like CML and pentosidine [102-104]. Nonetheless, ARBs could efficaciously delayed kidney failure progression [105,106], reduced the severity and risk of retinopathy in diabetic patients [107,108]. These findings support the use of ARBs for the management and prevention of diabetic vascular complications.
Various mechanisms have been proposed to explain ARB-facilitated AGE reduction. It is speculated that ARBs are potential PPARγ agonist that can exert glycaemic control and AGE-lowering effect similar to TZDs [109]. However, this is somewhat unlikely because ARB therapy has no apparent effect on fasting blood glucose and HbA¬1c level [101,110]. Another putative mechanism is by the restoration of glyoxalase-I activity. Basically, glyoxalase-I is a key enzyme in the detoxification of AGE precursors, namely glyoxal, methylglyoxal and 3-deoxyglucosone [111]. Candesartan was capable of rescuing glyoxalase-I mRNA expression and activity which were otherwise, impaired by angiotensin II [112]. Overexpression of glyoxalase I has been shown to cause a markedly low intracellular AGE level, suggesting that ARBs could promote detoxification of AGE precursors which reduces AGE accumulation [113]. Additionally, ARBs may also possess transition metal chelating ability which allows them to cease a number of oxidative reactions involved in the glycation pathway and dicarbonyl generation [114]. Collectively, these mechanisms could contribute to the AGE-lowering effect following ARB therapy.
Apart from AGE-lowering effect, ARB-treated diabetic patients showed decent improvements in various inflammatory and oxidative stress markers, namely, high-sensitive C-reactive protein, interleukins-6 and -18 [110,115]. This suggests possible inhibition of RAGE-associated cellular signaling as evidenced by the suppression of RAGE activation upon ARB treatment [116,117]. To date, the evidence about the inhibitory activity of ARBs on AGE-RAGE axis is fairly limited and so, such a bioactivity remains inconclusive. However, clinical studies did show beneficial effects of ARBs to retard diabetic microvascular disease progression which makes ARBs a desirable therapeutic candidate. Further investigations are necessary to delineate the possible underlying mechanisms of ARBs on AGE-RAGE axis.
Like ARBs, ACE inhibitors also act on RAAS. Instead of blocking the receptor, they inhibit the conversion of angiotensin I to angiotensin, thereby preventing the activation of angiotensin II type I receptor. One trial showed that ramipril could reduce AGE level [118] while another work demonstrated comparable therapeutic effect between enalapril and telmisartan in delaying diabetic nephropathy deterioration [106]. Like ARBs, it is proposed that ACE inhibitors can chelate the transition metals that catalyse AGE formation [114]. Nevertheless, the AGE-inhibiting effect of ACE inhibitors is still inconclusive due to inadequate evidence.
Another class of anti-hypertensive, CCBs including azelnidipine, amlodipine and nifedipine have also been tested for their AGE-RAGE inhibitory effect. Unlike ARBs and ACE inhibitors, CCBs do not target RAAS but instead, directly antagonize the calcium influx into muscle tissues to cause arterial dilation and decline in blood pressure [119]. Treatment with amlodipine failed to lower AGE level [102] whereas azelnidipine could [120]. Combined therapy with nifedipine and telmisartan significantly elevated sRAGE level, but whether or not such a beneficial effect was conferred by nifedipine is unknown [121]. CCBs are also found to repress RAGE expression in vitro by acting as PPARγ agonist [122]. However, clinically, such a RAGE modulatory effect of CCBs via PPARγ activation is unlikely as no impact on glycaemic parameters was observed [120]. On the other hand, AGE exposure has been shown to prolong calcium decay time in the cardiomyocytes which might induce abnormal contraction of the cardiac muscles [123]. Furthermore, CML-treated vascular smooth muscle cells had increased calcium release from the sarcoplasmic reticulum and calcium entry, contributing to the onset of enhanced contractility and hypertension [124]. The use of CCBs may be able to alleviate the adverse effects of AGE-RAGE axis on calcium regulation in muscle tissues. As such, like other antihypertensive agents, the inhibitory effects of CCBs on AGE-RAGE axis lack strong support from clinical evidence. Further clarifications on its mechanisms and efficacy are required to justify its clinical value in AGE-RAGE inhibition and managing diabetic complications (Table 4).
Vitamins B and its derivatives
Basically, the AGE-RAGE inhibitory effect of two types of vitamin B, namely vitamins B1 (thiamine and benfotiamine) and B6 (pyridoxine and pyridoxamine), have been examined. The clinical findings are summarized in Table 5. Some common pitfalls of the clinical trials are short treatment duration and small sample size. Generally, the studies reported mixed results on the AGE-RAGE inhibitory effect of vitamin B derivatives. Some studies detected beneficial AGE-lowering effect by benfotiamine [125,126] and by pyridoxamine [127] whereas others found no such activity [128-130].
Despite being in the same vitamin group, vitamins B1 and B6 are thought to have very distinct inhibitory mechanism on the AGE-RAGE axis. By supplying an essential cofactor of transketolase which is thiamine pyrophosphate, thiamine and benfotiamine enhance transketolase activity to channel fructose-6-phosphate and glyceraldehydes-3-phosphate from glycolysis into pentose phosphate pathway [131]. As this is also the rate-limiting step in pentose phosphate pathway, the activation of transketolase can effectively prevent the aggregation of the two metabolites which will otherwise be driven into dicarbonyl and AGE formation [132].
Conversely, pyridoxamine and pyridoxine can bind to catalytic redox metal ions which are vital to convert Amadori products to AGEs in the glycation pathway [133]. The post-amadori inhibition effectively prevents the formation of AGEs and hence, a new term "Amadorin" was coined to describe drugs behave similarly to vitamin B6 in the inhibition of AGE-RAGE axis [134]. However, the clinical prospect of both the vitamin B1 and B6 in alleviating the AGE-RAGE-mediated pathogenesis requires further investigation in light of the contradictions from the available studies (Table 5).
Other Investigational Interventions and Future Drug Development
In addition to the aforementioned five major classes of pharmacological agents, a few other drugs, including azeliragon (RAGE inhibitor), epalrestat (aldose reductase inhibitor), vitamin D and vitamin E, have also been tested clinically for their AGE-RAGE inhibitory effects as summarised in Table 6.
In this context, it is worth noting that RAGE-induced pathogenesis, particularly upon the interaction with amyloid β, is well-implicated in neurodegenerative diseases like Alzheimer's disease [135]. Therefore, azeliragon or PF-04494700 or TTP488, which is a small-molecule RAGE antagonist, has been developed as an investigational drug against Alzheimer's disease and diabetic neuropathy. Unfortunately, the development of the drug for the latter has been discontinued. For Alzheimer's disease, a preclinical study using transgenic mice that overexpressed human amyloid precursor proteins demonstrated an excellent, dose-dependent therapeutic efficacy of azeliragon in terms of the amyloid plaque formation, proinflammatory response, cerebral glucose utilisation and behavioural impairment [136]. However, in human clinical trials, at higher dosages (≥ 20 mg/day), the drug seemed to accelerate cognitive dysfunction [137]. Low-dose regimen, on the other hand, might confer some protection against cognitive deterioration, notably among the patients with mild Alzheimer's disease [138]. Currently, two phase 3 clinical studies are on-going to explore the short- and long-term efficacy and safety of azeliragon. Positive findings from these trials may support the use of azeliragon in diabetic vasculopathy.
Next, as mentioned previously, reactive carbonyl species which is AGE precursors can be derived from the polyol pathway. Therefore, blocking the pathway is an attractive choice to reduce AGE formation. In fact, the reduction of dicarbonyl compounds like CML and 3-deoxyglucosone with the use of epalrestat which is an aldose reductase inhibitor, is strongly supported by clinical evidence [139,140]. The inhibition of aldose reductase and blockade of polyol pathway is further translated into other beneficial effects, most notably the delayed progression of diabetic retinopathy, nephropathy and neuropathy [141,142]. Thus far, the clinical results of aldose reductase inhibitor, especially in diabetic cardiovascular autonomic neuropathy, are exceedingly favourable [143], making it a valuable drug for the therapy of diabetic complications.
The inhibitory effect of vitamins D and E on AGE-RAGE axis have also been examined in non-diabetic patients, both of which showed a certain extent of beneficial effects by increasing sRAGE or lowering circulating AGEs respectively. The actual mechanism remains unclear, but may be partially explained by their antioxidant properties [144,145]. Speaking of antioxidant activity, α-lipoic acid which is a potent antioxidant can also confer AGE-lowering effect in diabetic patients when it is used together with other potential AGE inhibitors like benfotiamine and pyridoxine [126,146]. However, these studies did not include an "α-lipoic acid only" cohort and hence, its effect on AGE-RAGE axis independent from the interaction with other compounds is unclear. It is worth mentioning that α-lipoic acid has been extensively tested in diabetic patients and consistently shown to alleviate oxidative stress, improve lipid and glucose homeostasis and most importantly, enhance the peripheral neurological function in patients with diabetic polyneuropathy [147,148]. This points to a promising clinical prospect of α-lipoic acid in diabetes and its related vasculopathy.
In regard to the development of specific RAGE inhibitors, increasing efforts and resources are channelled into synthetic medicine. One of the earlier examples is FPS-ZM1, which is a high-affinity synthetic RAGE inhibitor that binds to the V domain of the receptor [149]. FPS-ZM1 also readily crosses the blood-brain-barrier, inhibits β-secretase and production of amyloid β peptide, making it a potential therapy for Alzheimer's disease [149]. Han, et al. also reported successful synthesis of a 2-aminopyrimidine-based small molecule that can inhibit ligand-RAGE interaction and confers similar therapeutic effects to FPS-ZM1 in transgenic mouse models with Alzheimer's disease [150]. With the assistance of molecular docking, the small molecule was also predicted to bind to the V-domain of RAGE. In fact, with the advancement in computational technology, it is entirely possible to screen a wide arrays of molecular structures for promising RAGE inhibitors with well-designed algorithms. For instance, one such molecule, Compound 62, has been identified to demonstrate excellent bioavailability, minimal mutagenicity and carcinogenicity besides having the highest affinity to RAGE compared to many pre-existing specific RAGE inhibitors, all with the use of computational tools [151]. Even though further experimental validation is necessary, it is undeniable that computational advancement could significantly revolutionise not only the AGE-RAGE and diabetes research, but also many other aspects in medicine and drug development in the near future (Table 6).
`Conclusion
As the AGE-RAGE axis is reckoned to be one of the major driving forces to the onset of diabetic vascular complications, potent AGE-RAGE inhibitory agents are therefore, highly sought after. However, aside from glucose- and lipid-lowering medications, the clinical studies of most AGE-RAGE inhibitors yielded mixed results in terms of their clinical efficacy and practicality. This is strongly indicative of a few arguments: (1) Current understanding of AGE-RAGE-mediated pathogenesis is incomplete; (2) Inhibition of AGE-RAGE axis may be better as adjunctive instead or primary therapy; (3) RAGE ligands other than AGE could play a predominant role in RAGE-mediated signal transduction; (4) The clinical trials were not empowered to detect minor beneficial effects. In light of the complex pathogenesis and multifactorial nature of diabetes- and cardiovascular-associated comorbidities, such mixed and confusing clinical findings are not entirely surprisingly. However, this does not imply that targeting AGE-RAGE axis is a futile attempt. In fact, recent breakthrough in Alzheimer's disease with the use of specific RAGE inhibitor (azeliragon) points out that blockade of RAGE could indeed confer positive impacts, particularly at the early stage of the disease. Likewise, targeting AGE-RAGE axis during pre-diabetic state may help to slow down the progression of diabetic vascular complications. As for currently available drugs, metformin, TZDs and statins show great potential to be developed as AGE-RAGE inhibitors as supported by concrete clinical evidence. Aldose reductase inhibitors are also promising candidates but the clinical evidence on AGE-RAGE axis is limited. As such, AGE-RAGE antagonists remain as an interesting clinical option for the treatment of diabetes-associated complications. Despite the contradictory results, exploratory studies that identify the specific patient populations whom will be benefited from the treatment is highly recommended.
Acknowledgements
The work was supported the School of Science, Monash University Malaysia.
Conflicts of Interest
None.
References
- World Health Organization (2009) Global health risks: Mortality and burden of disease attributable to selected major risks. Geneva, Switzerland.
- World Health Organization (2016) Global report on diabetes. Geneva, Switzerland.
- Singh R, Barden A, Mori T, et al. (2001) Advanced glycation end-products: a review. Diabetologia 44: 129-146.
- Kilhovd BK, Berg TJ, Birkeland KI, et al. (1999) Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care 22: 1543-1548.
- Turk Z (2010) Glycotoxines, carbonyl stress and relevance to diabetes and its complications. Physiol Res 59: 147-156.
- Dunlop M (2000) Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int Suppl 77: S3-S12.
- Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344: 109-116.
- Fu MX, Requena JR, Jenkins AJ, et al. (1996) The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 271: 9982-9986.
- Miyata T, Ueda Y, Yamada Y, et al. (1998) Accumulation of carbonyls accelerates the formation of pentosidine, an advanced glycation end product: carbonyl stress in uremia. J Am Soc Nephrol 9: 2349-2356.
- Ahmed N (2005) Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes Res Clin Pract 67: 3-21.
- Goldin A, Beckman JA, Schmidt AM, et al. (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114: 597-605.
- Sims TJ, Rasmussen LM, Oxlund H, et al. (1996) The role of glycation cross-links in diabetic vascular stiffening. Diabetologia 39: 946-951.
- Ma H, Li SY, Xu P, et al. (2009) Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med 13: 1751-1764.
- Xie J, Méndez JD, Méndez-Valenzuela V, et al. (2013) Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal 25: 2185-2197.
- Bierhaus A, Schiekofer S, Schwaninger M, et al. (2001) Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 50: 2792-2808.
- Semba RD, Nicklett EJ, Ferrucci L (2010) Does accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol A Biol Sci Med Sci 65: 963-975.
- Li H, Nakamura S, Miyazaki S, et al. (2006) N2-carboxyethyl-2′-deoxyguanosine, a DNA glycation marker, in kidneys and aortas of diabetic and uremic patients. Kidney Int 69: 388-392.
- Waris S, Winklhofer-Roob BM, Roob JM, et al. (2015) Increased DNA dicarbonyl glycation and oxidation markers in patients with type 2 diabetes and link to diabetic nephropathy. J Diabetes Res 2015: 915486.
- Barnaby OS, Cerny RL, Clarke W, et al. (2011) Comparison of modification sites formed on human serum albumin at various stages of glycation. Clin Chim Acta 412: 277-285.
- Rubenstein DA, Yin W (2009) Glycated albumin modulates platelet susceptibility to flow induced activation and aggregation. Platelets 20: 206-215.
- Dutta U, Cohenford MA, Dain JA (2006) Monitoring the effect of glucosamine and glyceraldehyde glycation on the secondary structure of human serum albumin and immunoglobulin G: An analysis based on circular dichroism, thermal melting profiles and UV-fluorescence spectroscopy. Anal Chim Acta 558: 187-194.
- Jairajpuri DS, Fatima S, Saleemuddin M (2007) Immunoglobulin glycation with fructose: a comparative study. Clin Chim Acta 378: 86-92.
- Hammes HP, Weiss A, Hess S, et al. (1996) Modification of vitronectin by advanced glycation alters functional properties in vitro and in the diabetic retina. Lab Invest 75: 325-338.
- Tanaka S, Avigad G, Brodsky B, et al. (1988) Glycation induces expansion of the molecular packing of collagen. J Mol Biol 203: 495-505.
- Howard EW, Benton R, Ahern-Moore J, et al. (1996) Cellular contraction of collagen lattices is inhibited by nonenzymatic glycation. Exp Cell Res 228: 132-137.
- Charonis AS, Reger LA, Dege JE, et al. (1990) Laminin alterations after in vitro nonenzymatic glycosylation. Diabetes 39: 807-814.
- Aronson D (2003) Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens 21: 3-12.
- Rabbani N, Godfrey L, Xue M, et al. (2011) Glycation of LDL by methylglyoxal increases arterial atherogenicity: a possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 60: 1973-1980.
- Jinnouchi Y, Sano H, Nagai R, et al. (1998) Glycolaldehyde-modified low density lipoprotein leads macrophages to foam cells via the macrophage scavenger receptor. J Biochem 123: 1208-1217.
- Braach N, Frommhold D, Buschmann K, et al. (2014) RAGE controls activation and anti-inflammatory signalling of protein C. PLoS One 9: e89422.
- Schmidt AM, Hori O, Chen JX, et al. (1995) Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 96: 1395-1403.
- Kim I, Moon SO, Park SK, et al. (2001) Angiopoietin-1 reduces VEGF-stimulated leukocyte adhesion to endothelial cells by reducing ICAM-1, VCAM-1, and E-selectin expression. Circ Res 89: 477-479.
- Neumann A, Schinzel R, Palm D, et al. (1999) High molecular weight hyaluronic acid inhibits advanced glycation endproduct-induced NF-kappaB activation and cytokine expression. FEBS Lett 453: 283-287.
- Wendt TM, Tanji N, Guo J, et al. (2003) RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 162: 1123-1137.
- Choudhuri S, Chowdhury IH, Das S, et al. (2015) Role of NF-κB activation and VEGF gene polymorphisms in VEGF up regulation in non-proliferative and proliferative diabetic retinopathy. Mol Cell Biochem 405: 265-279.
- Harja E, Bu DX, Hudson BI, et al. (2008) Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest 118: 183-194.
- Quehenberger P, Bierhaus A, Fasching P, et al. (2000) Endothelin 1 transcription is controlled by nuclear factor-kappaB in AGE-stimulated cultured endothelial cells. Diabetes 49: 1561-1570.
- Rojas A, Romay S, González D, et al. (2000) Regulation of endothelial nitric oxide synthase expression by albumin-derived advanced glycosylation end products. Circ Res 86: E50-E54.
- Xu B, Chibber R, Ruggiero D, et al. (2003) Impairment of vascular endothelial nitric oxide synthase activity by advanced glycation end products. FASEB J 17: 1289-1291.
- Bucciarelli LG, Ananthakrishnan R, Hwang YC, et al. (2008) RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes 57: 1941-1951.
- Nukada H (2014) Ischemia and diabetic neuropathy. Handb Clin Neurol 126: 469-487.
- Kamide T, Kitao Y, Takeichi T, et al. (2012) RAGE mediates vascular injury and inflammation after global cerebral ischemia. Neurochem Int 60: 220-228.
- Thornalley PJ (2003) Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys 419: 31-40.
- Itakura M, Yoshikawa H, Bannai C, et al. (1991) Aminoguanidine decreases urinary albumin and high-molecular-weight proteins in diabetic rats. Life Sci 49: 889-897.
- Hammes HP, Martin S, Federlin K, et al. (1991) Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A 88: 11555-11558.
- Bolton WK, Cattran DC, Williams ME, et al. (2004) Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol 24: 32-40.
- Freedman BI, Wuerth JP, Cartwright K, et al. (1999) Design and baseline characteristics for the aminoguanidine clinical trial in overt type 2 diabetic nephropathy (ACTION II). Control Clin Trials 20: 493-510.
- Nilsson BO (1999) Biological effects of aminoguanidine: an update. Inflamm Res 48: 509-515.
- Hess EV (2002) Environmental chemicals and autoimmune disease: cause and effect. Toxicology 181-182: 65-70.
- Kass DA, Shapiro EP, Kawaguchi M, et al. (2001) Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation 104: 1464-1470.
- Fujimoto N, Hastings JL, Carrick-Ranson G, et al. (2013) Cardiovascular effects of 1 year of alagebrium and endurance exercise training in healthy older individuals. Circ Heart Fail 6: 1155-1164.
- Hartog JWL, Willemsen S, van Veldhuisen DJ, et al. (2011) Effects of alagebrium, an advanced glycation endproduct breaker, on exercise tolerance and cardiac function in patients with chronic heart failure. Eur J Heart Fail 13: 899-908.
- Oudegeest-Sander MH, Rikkert MGMO, Smits P, et al. (2013) The effect of an advanced glycation end-product crosslink breaker and exercise training on vascular function in older individuals: A randomized factorial design trial. Exp Gerontol 48: 1509-1517.
- Carrick-Ranson G, Fujimoto N, Shafer KM, et al. (2016) The effect of 1 year of alagebrium and moderate-intensity exercise training on left ventricular function during exercise in seniors: a randomized controlled trial. J Appl Physiol 121: 528-536.
- Little WC, Zile MR, Kitzman DW, et al. (2005) The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. J Card Fail 11: 191-195.
- Zieman SJ, Melenovsky V, Clattenburg L, et al. (2007) Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J Hypertens 25: 577-583.
- Vaitkevicius PV, Lane M, Spurgeon H, et al. (2001) A cross-link breaker has sustained effects on arterial and ventricular properties in older rhesus monkeys. Proc Natl Acad Sci U S A 98: 1171-1175.
- Chandra KP, Shiwalkar A, Kotecha J, et al. (2009) Phase I clinical studies of the advanced glycation end-product (AGE)-breaker TRC4186. Clin Drug Investig 29: 559-575.
- Kiho T, Kato M, Usui S, et al. (2005) Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin Chim Acta 358: 139-145.
- Beisswenger PJ, Howell SK, Touchette AD, et al. (1999) Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 48: 198-202.
- Chakraborty A, Chowdhury S, Bhattacharyya M (2011) Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res Clin Pract 93: 56-62.
- Esteghamati A, Eskandari D, Mirmiranpour H, et al. (2013) Effects of metformin on markers of oxidative stress and antioxidant reserve in patients with newly diagnosed type 2 diabetes: A randomized clinical trial. Clin Nutr 32: 179-185.
- Lund SS, Tarnow L, Stehouwer CD, et al. (2008) Impact of metformin versus repaglinide on non-glycaemic cardiovascular risk markers related to inflammation and endothelial dysfunction in non-obese patients with type 2 diabetes. Eur J Endocrinol 158: 631-641.
- Mirmiranpour H, Mousavizadeh M, Noshad S, et al. (2013) Comparative effects of pioglitazone and metformin on oxidative stress markers in newly diagnosed type 2 diabetes patients: A randomized clinical trial. J Diabetes Complications 27: 501-507.
- Spiegelman BM (1998) PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes 47: 507-514.
- Wang K, Zhou Z, Zhang M, et al. (2006) Peroxisome proliferator-activated receptor γ down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther 317: 37-43.
- Marx N, Walcher D, Ivanova N, et al. (2004) Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes 53: 2662-2668.
- Tan KC, Chow WS, Tso AW, et al. (2007) Thiazolidinedione increases serum soluble receptor for advanced glycation end-products in type 2 diabetes. Diabetologia 50: 1819-1825.
- Oz Gul O, Tuncel E, Yilmaz Y, et al. (2010) Comparative effects of pioglitazone and rosiglitazone on plasma levels of soluble receptor for advanced glycation end products in type 2 diabetes mellitus patients. Metabolism 59: 64-69.
- Raucci A, Cugusi S, Antonelli A, et al. (2008) A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 22: 3716-3727.
- Park IH, Yeon SI, Youn JH, et al. (2004) Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol Immunol 40: 1203-1211.
- Schmidt AM (2015) Soluble RAGEs - Prospects for treating & tracking metabolic and inflammatory disease. Vascul Pharmacol 72: 1-8.
- Tam Xystus HL, Shiu Sammy WM, Leng L, et al. (2011) Enhanced expression of receptor for advanced glycation end-products is associated with low circulating soluble isoforms of the receptor in Type 2 diabetes. Clin Sci (Lond) 120: 81-89.
- Sakata K, Hayakawa M, Yano Y, et al. (2013) Efficacy of alogliptin, a dipeptidyl peptidase-4 inhibitor, on glucose parameters, the activity of the advanced glycation end product (AGE) - receptor for AGE (RAGE) axis and albuminuria in Japanese type 2 diabetes. Diabetes Metab Res Rev 29: 624-630.
- Koyama H, Tanaka S, Monden M, et al. (2014) Comparison of effects of pioglitazone and glimepiride on plasma soluble RAGE and RAGE expression in peripheral mononuclear cells in type 2 diabetes: Randomized controlled trial (PioRAGE). Atherosclerosis 234: 329-334.
- Bonetti PO, Lerman LO, Napoli C, et al. (2003) Statin effects beyond lipid lowering-are they clinically relevant? Eur Heart J 24: 225-248.
- Jinnouchi Y, Yamagishi S, Takeuchi M, et al. (2006) Atorvastatin decreases serum levels of advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin Exp Med 6: 191-193.
- Santilli F, Bucciarelli L, Noto D, et al. (2007) Decreased plasma soluble RAGE in patients with hypercholesterolemia: Effects of statins. Free Radic Biol Med 43: 1255-1262.
- Kimura Y, Hyogo H, Yamagishi S, et al. (2010) Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J Gastroenterol 45: 750-757.
- Tam HL, Shiu SW, Wong Y, et al. (2010) Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis 209: 173-177.
- Fukushima Y, Daida H, Morimoto T, et al. (2013) Relationship between advanced glycation end products and plaque progression in patients with acute coronary syndrome: The JAPAN-ACS Sub-study. Cardiovasc Diabetol 12: 5.
- Cuccurullo C, Iezzi A, Fazia ML, et al. (2006) Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler Thromb Vasc Biol 26: 2716-2723.
- Quade-Lyssy P, Kanarek AM, Baiersdörfer M, et al. (2013) Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. J Lipid Res 54: 3052-3061.
- Matthews V, Schuster B, Schütze S, et al. (2003) Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J Biol Chem 278: 38829-38839.
- Kojro E, Gimpl G, Lammich S, et al. (2001) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc Natl Acad Sci U S A 98: 5815-5820.
- Murai T, Maruyama Y, Mio K, et al. (2011) Low cholesterol triggers membrane microdomain-dependent CD44 shedding and suppresses tumor cell migration. J Biol Chem 286: 1999-2007.
- Vistoli G, De Maddis D, Cipak A, et al. (2013) Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic Res 47: 3-27.
- Wagner AH, Köhler T, Rückschloss U, et al. (2000) Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol 20: 61-69.
- Wassmann S, Laufs U, Bäumer AT, et al. (2001) HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 37: 1450-1457.
- Wassmann S, Laufs U, Müller K, et al. (2002) Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol 22: 300-305.
- Yoon SJ, Yoon YW, Lee BK, et al. (2009) Potential role of HMG CoA reductase inhibitor on oxidative stress induced by advanced glycation endproducts in vascular smooth muscle cells of diabetic vasculopathy. Exp Mol Med 41: 802-811.
- Tomizawa A, Hattori Y, Inoue T, et al. (2011) Fenofibrate suppresses microvascular inflammation and apoptosis through adenosine monophosphate-activated protein kinase activation. Metabolism 60: 513-522.
- Calkin AC, Cooper ME, Jandeleit-Dahm KA, et al. (2006) Gemfibrozil decreases atherosclerosis in experimental diabetes in association with a reduction in oxidative stress and inflammation. Diabetologia 49: 766-774.
- Keech A, Simes RJ, Barter P, et al. (2005) Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366: 1849-1861.
- The ACCORD Study Group, ACCORD Eye Study Group, Chew EY, et al. (2010) Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 363: 233-244.
- Shimomura M, Oyama J, Takeuchi M, et al. (2016) Acute effects of statin on reduction of angiopoietin-like 2 and glyceraldehyde-derived advanced glycation end-products levels in patients with acute myocardial infarction: a message from SAMIT (Statin for Acute Myocardial Infarction Trial). Heart Vessels 31: 1583-1589.
- Tsuyoshi Nozue, Sho-ichi Yamagishi, Masayoshi Takeuchi, et al. (2014) Effect of statins on the serum soluble form of receptor for advanced glycation end-products and its association with coronary atherosclerosis in patients with angina pectoris. IJC Metabolic and Endocrine 4: 47-52.
- Zaman MA, Oparil S, Calhoun DA (2002) Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov 1: 621-636.
- Monacelli F, Poggi A, Storace D, et al. (2006) Effects of valsartan therapy on protein glycoxidation. Metabolism 55: 1619-1624.
- Honda H, Hosaka N, Aoshima Y, et al. (2012) Olmesartan medoxomil is associated with decreased plasma AGEs, pentosidine, and N-(Epsilon)-carboxymethyl-lysine levels in hemodialysis patients. Clin Exp Hypertens 34: 17-23.
- Saisho Y, Komiya N, Hirose H (2006) Effect of valsartan, an angiotensin II receptor blocker, on markers of oxidation and glycation in Japanese type 2 diabetic subjects: Blood pressure-independent effect of valsartan. Diabetes Res Clin Pract 74: 201-203.
- Busch M, Franke S, Wolf G, et al. (2008) Serum levels of the advanced glycation end products Nε-carboxymethyllysine and pentosidine are not influenced by treatment with the angiotensin receptor II type 1 blocker irbesartan in patients with type 2 diabetic nephropathy and hypertension. Nephron Clin Pract 108: 291-297.
- Engelen L, Persson F, Ferreira I, et al. (2011) Irbesartan treatment does not influence plasma levels of the advanced glycation end products Nε (1-carboxymethyl) lysine and Nε (1-carboxyethyl)lysine in patients with type 2 diabetes and microalbuminuria. A randomized controlled trial. Nephrol Dial Transplant 26: 3573-3577.
- Hartog JW, van de Wal RM, Schalkwijk CG, et al. (2010) Advanced glycation end-products, anti-hypertensive treatment and diastolic function in patients with hypertension and diastolic dysfunction. Eur J Heart Fail 12: 397-403.
- Lewis EJ, Hunsicker LG, Clarke WR, et al. (2001) Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345: 851-860.
- Barnett AH, Bain SC, Bouter P, et al. (2004) Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 351: 1952-1961.
- Sjølie AK, Klein R, Porta M, et al. (2008) Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT-Protect 2): a randomised placebo-controlled trial. Lancet 372: 1385-1393.
- Chaturvedi N, Porta M, Klein R, et al. (2008) Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials. Lancet 372: 1394-1402.
- Schupp M, Janke J, Clasen R, et al. (2004) Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-γ activity. Circulation 109: 2054-2057.
- Kuboki K, Iso K, Murakami E, et al. (2007) Effects of valsartan on inflammatory and oxidative stress markers in hypertensive, hyperglycemic patients: An open-label, prospective study. Curr Ther Res Clin Exp 68: 338-348.
- Thornalley PJ (2003) Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans 31: 1343-1348.
- Miller AG, Tan G, Binger KJ, et al. (2010) Candesartan attenuates diabetic retinal vascular pathology by restoring glyoxalase-I function. Diabetes 59: 3208-3215.
- Shinohara M, Thornalley PJ, Giardino I, et al. (1998) Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest 101: 1142-1147.
- Miyata T, van Ypersele de Strihou C, Ueda Y, et al. (2002) Angiotensin II receptor antagonists and angiotensin-converting enzyme inhibitors lower in vitro the formation of advanced glycation end products: Biochemical mechanisms. J Am Soc Nephrol 13: 2478-2487.
- Persson F, Rossing P, Hovind P, et al. (2006) Irbesartan treatment reduces biomarkers of inflammatory activity in patients with type 2 diabetes and microalbuminuria: An IRMA 2 substudy. Diabetes 55: 3550-3555.
- Fujita M, Okuda H, Tsukamoto O, et al. (2006) Blockade of angiotensin II receptors reduces the expression of receptors for advanced glycation end products in human endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology 26: e138-e139.
- Kikuchi K, Tancharoen S, Ito T, et al. (2013) Potential of the angiotensin receptor blockers (ARBs) telmisartan, irbesartan, and candesartan for inhibiting the HMGB1/RAGE axis in prevention and acute treatment of stroke. Int J Mol Sci 14: 18899-18924.
- Sebekova K, Gazdikova K, Syrova D, et al. (2003) Effects of ramipril in nondiabetic nephropathy: improved parameters of oxidatives stress and potential modulation of advanced glycation end products. J Hum Hypertens 17: 265-270.
- DeWitt CR, Waksman JC (2004) Pharmacology, pathophysiology and management of calcium channel blocker and beta-blocker toxicity. Toxicol Rev 23: 223-238.
- Nakamura T, Sato E, Fujiwara N, et al. (2011) Calcium channel blocker inhibition of AGE and RAGE axis limits renal injury in nondiabetic patients with stage I or II chronic kidney disease. Clin Cardiol 34: 372-377.
- Falcone C, Buzzi MP, Bozzini S, et al. (2012) Microalbuminuria and sRAGE in high-risk hypertensive patients treated with nifedipine/telmisartan combination treatment: A substudy of TALENT. Mediators of Inflammation 2012: 1-6.
- Matsui T, Yamagishi S, Takeuchi M, et al. (2009) Nifedipine, a calcium channel blocker, inhibits advanced glycation end product (AGE)-elicited mesangial cell damage by suppressing AGE receptor (RAGE) expression via peroxisome proliferator-activated receptor-gamma activation. Biochemical and Biophysical Research Communications 385: 269-272.
- Petrova R, Yamamoto Y, Muraki K, et al. (2002) Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J Mol Cell Cardiol 34: 1425-1431.
- Simard E, Söllradl T, Maltais JS, et al. (2015) Receptor for advanced glycation end-products signaling interferes with the vascular smooth muscle cell contractile phenotype and function. PLoS One 10: e0128881.
- Stirban A, Negrean M, Stratmann B, et al. (2006) Benfotiamine prevents macro-and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 29: 2064-2071.
- Du X, Edelstein D, Brownlee M (2008) Oral benfotiamine plus α-lipoic acid normalises complication-causing pathways in type 1 diabetes. Diabetologia 51: 1930-1932.
- Williams ME, Bolton WK, Khalifah RG, et al. (2007) Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol 27: 605-614.
- Alkhalaf A, Klooster A, van Oeveren W, et al. (2010) A double-blind, randomized, placebo-controlled clinical trial on benfotiamine treatment in patients with diabetic nephropathy. Diabetes Care 33: 1598-1601.
- Alkhalaf A, Kleefstra N, Groenier KH, et al. (2012) Effect of benfotiamine on advanced glycation endproducts and markers of endothelial dysfunction and inflammation in diabetic nephropathy. PLoS One 7: e40427.
- Nascimento MM, Suliman ME, Murayama Y, et al. (2006) Effect of high-dose thiamine and pyridoxine on advanced glycation end products and other oxidative stress markers in hemodialysis patients: A randomized placebo-controlled study. J Ren Nutr 16: 119-124.
- Hammes HP, Du X, Edelstein D, et al. (2003) Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 9: 294-299.
- Babaei-Jadidi R, Karachalias N, Ahmed N, et al. (2003) Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes 52: 2110-2120.
- Voziyan PA, Khalifah RG, Thibaudeau C, et al. (2003) Modification of proteins in vitro by physiological levels of glucose: Pyridoxamine inhibits conversion of amadori intermediate to advnaced glycation end-products through binding of redox metal ions. J Biol Chem 278: 46616-46624.
- Khalifah RG, Baynes JW, Hudson BG (1999) Amadorins: Novel post-amadori inhibitors of advanced glycation reactions. Biochem Biophys Res Commun 257: 251-258.
- Cai Z, Liu N, Wang C, et al. (2016) Role of RAGE in Alzheimer's disease. Cell Mol Neurobiol 36: 483-495.
- Kostura MJ, Kindy MS, Burstein A, et al. (2014) Efficacy of RAGE antagonists in murine model of Alzheimer's disease. Alzheimers & Dementia 10: 638-639.
- Galasko D, Bell J, Mancuso JY, et al. (2014) Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurology 82: 1536-1542.
- Burstein AH, Grimes I, Galasko DR, et al. (2014) Effect of TTP488 in patients with mild to moderate Alzheimer's disease. BMC Neurol 14: 12.
- Hamada Y, Nakamura J, Naruse K, et al. (2000) Epalrestat, an aldose reductase ihibitor, reduces the levels of Nepsilon-(carboxymethyl) lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diabetes Care 23: 1539-1544.
- Kawai T, Takei I, Tokui M, et al. (2010) Effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy in patients with type 2 diabetes, in relation to suppression of Nɛ-carboxymethyl lysine. J Diabetes Complications 24: 424-432.
- Hotta N, Akanuma Y, Kawamori R, et al. (2006) Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: The 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes Care 29: 1538-1544.
- Hotta N, Kawamori R, Fukuda M, et al. (2012) Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabet Med 29: 1529-1533.
- Hu X, Li S, Yang G, et al. (2014) Efficacy and safety of aldose reductase inhibitor for the treatment of diabetic cardiovascular autonomic neuropathy: Systematic review and meta-analysis. PLoS One 9: e87096.
- Salum E, Kals J, Kampus P, et al. (2013) Vitamin D reduces deposition of advanced glycation end-products in the aortic wall and systemic oxidative stress in diabetic rats. Diabetes Res Clin Pract 100: 243-249.
- Reaven PD, Herold DA, Barnett J, et al. (1995) Effects of vitamin E on susceptibility of low-density lipoprotein and low-density lipoprotein subfractions to oxidation and on protein glycation in NIDDM. Diabetes Care 18: 807-816.
- Noori N, Tabibi H, Hosseinpanah F, et al. (2013) Effects of combined lipoic acid and pyridoxine on albuminuria, advanced glycation end-products, and blood pressure in diabetic nephropathy. Int J Vitam Nutr Res 83: 77-85.
- Ziegler D, Nowak H, Kempler P, et al. (2004) Treatment of symptomatic diabetic polyneuropathy with the antioxidant α-lipoic acid: a meta-analysis. Diabet Med 21: 114-121.
- Saeid Golbidi, Mohammad Badran, Ismail Laher (2011) Diabetes and alpha lipoic acid. Front Pharmacol 2: 69.
- Deane R, Singh I, Sagare AP, et al. (2012) A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 122: 1377-1392.
- Han YT, Choi GI, Son D, et al. (2012) Ligand-based design, synthesis, and biological evaluation of 2-aminopyrimidines, a novel series of receptor for advanced glycation end products (RAGE) inhibitors. J Med Chem 55: 9120-9135.
- Devi Alaparthi M, Gopinath G, Bandaru S, et al. (2016) Virtual screening of RAGE inhibitors using molecular docking. Bioinformation 12: 124-130.
- Sabbagh MN, Agro A, Bell J, et al. (2011) PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord 25: 206-212.
- Irani M, Minkoff H, Seifer DB, et al. (2014) Vitamin D increases serum levels of the soluble receptor for advanced glycation end products in women with PCOS. J Clin Endocrinol Metab 99: E886-E890.
- Baragetti I, Furiani S, Vettoretti S, et al. (2006) Role of vitamin E-coated membrane in reducing advanced glycation end products in hemodialysis patients: A pilot study. Blood Purif 24: 369-376.
Corresponding Author
Hong Sheng Cheng, School of Science, Monash University Malaysia, 46150 Bandar Sunway, Selangor, Malaysia, Tel: +6010-933-7034, Fax: +603-5514-6099.
Copyright
© 2017 Cheng HS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.