Loss of Oligodendrocytes by Oxidative Phosphorylation Inhibitor Rotenone and its Reversal by Phenylbutyrate (PB) in Human Brain Developmental Organoid Model
Abstract
Brain oligodendrocytes have been shown to be particularly affected by hypoxia and mitochondrial insufficiency. To model their effects on human brain development, we exposed developing human iPSC derived cerebral organoids to rotenone, a mitochondrial oxidative phosphorylation (OXPHOS) inhibitor. A short-term, 3-day, rotenone exposure depleted proliferating (Ki67+) organoid neural progenitor cells (NPC) while increasing distances between the residual NPC. Rotenone treatment also depleted the population of O4+ oligodendrocytes and a similar trend was observed with the double stained Ki67+/O4+ oligodendrocyte progenitor cells (OPC). When allowed an additional 10-days post- rotenone recovery time, the organoids showed spontaneous partial restoration of the O4+ population. This regeneration was significantly enhanced by treatment with phenylbutyrate (PB) known to stimulate acetyl-CoA and energy production from glucose by increasing activity of the mitochondrial Pyruvate Dehydrogenase. Specifically, PB stimulated the OPC numbers and increased the O4+ oligodendrocyte population depleted by 3-day Rotenone but it did not affect the Ki67+ NPC population. A longer, 6-day rotenone exposure lead to an extensive loss of NPC, OPC and O4+ oligodendrocytes. Neither the spontaneous nor PB-induced recoveries of NPC, OPC and O4+ oligodendrocytes were noted. In summary, following a transient oxidative insult, the oligodendrocyte population is capable of significant spontaneous regeneration and PB may be used to enhance the recovery. The cerebral organoids provide an effective model for studying human brain developmental oligodendrogenesis, its disruption by oxidative stress and for development of new corrective therapies.
Keywords
Hypoxia, Oxidative phosphorylation, Rotenone, Oligdendrogenesis, Cerebral organoids, Phenylvutyrate
Introduction
The efficiency and speed of neuronal signal transmission in CNS these signals is largely reliant upon myelin. Within the CNS, myelin is formed by oligodendrocytes [1-3]. The OPC are generated from multipotent NSC lining the CNS ventricles. The life of Oligodendrocytes has four distinct phases. The first is the birth, migration, and proliferation of oligodendrocyte progenitor cells (OPC), followed by morphological differentiation in the second phase in which the oligodendrocytes establish an expansive network of processes. In the third phase, axonal contact leads to ensheathment and generation of compact myelin around target axons, and in the last phase mature oligodendrocytes provide long-term trophic and metabolic support of the encased axon [2].
During development, the Neuro-Epithelial Cells (NECs) give rise to the primary stem- cells (NSC), the apical radial glia (aRG) [4]. As the brain develops, aRGs will become confined to the primary cortical germinal layer, known as the ventricular zone (VZ) and subsequently to subventricular zone. The radial fiber provides scaffold on which progenitor cells migrate basally outward [4,5]. aRGs have the capability of generating neurons, astrocytes, and oligodendrocytes or intermediate transient amplifying populations, progenitor cells which will later differentiate [4,6-9]. A new population of the radial glia, basal (b) RG migrate basally to the second germinal layer the SVZ where they undergo mostly symmetric neurogenic divisions, producing the vast majority of neurons in all layers of the cerebral cortex [4,5,10,11]. During the middle, neurogenic, embryonic stage, intermediate progenitors migrate on the long radial fibers from the VZ or SVZ through a cell-poor intermediate zone (IZ) to their final destination within the outer cortical plate (CP). The subsequent waves of neurons position themselves in an inside-out manner with the deepest cortical layers representing earlier-born neurons while more superficial layers are composed of later-born neurons [5]. Later in the third stage the intermediate progenitors can become astrocytes or OPCs [12].
Oligodendrocytes develop through waves of OPC production and migration initially from the medial ganglionic eminence and under the transcriptional control of gene Nkx2.1 [13-15]. These OPCs populate the entire embryonic telencephalon including the cerebral cortex [16]. As development progresses, a second wave of OPCs emanating from the lateral and caudal ganglionic eminences under the transcriptional control of gene Gsh2, migrate dorsally to the cortex and disperse throughout the forebrain [16,17]. In mouse brain, the third and final wave occurs after birth, embryonic day 21/postnatal day 0, which gives rise to the majority of adult oligodendrocytes and is under the control of transcription factor Emx1. These OPCs project radially from the dorsal and outer SVZ, directly next to the developing corpus callosum where they spread out in order to populate most of the future brain [16].
While most of the scientific understanding of oligodendrogenesis come from rodent studies, analysis of human fetal brain tissue has revealed that the first OPCs emerge in the developing brain at 10 weeks gestation, followed by an expansion of the population before mid-gestation (15-20 weeks) [18-20]. Similar to the rodent brain, OPCs in the human forebrain are derived from the lateral-medial ganglionic eminence and the SVZ, where they migrate towards the cortical plate [21,22].
Further evidence of OPC heterogeneity in the human brain comes from Leong, et al. [23] who demonstrated the presence of different OPC subtypes based on marker expression (A2B5, O4, and MOG) in both fetal (Trimesters 1 and 2) and adult brain tissue using fluorescence activated cell sorting [23]. Expression of miRNA in fetal OPC is higher than in adult brain OPC, suggesting that the human brain also contains different subpopulations of OPCs, originating from different brain areas [24].
Brain oligodendrocytes have been shown to be particularly affected by hypoxia and mitochondrial insufficiency (impaired mitochondrial oxidative phosphorylation, OXPHOS) [25,26]. Studies have shown high oligodendrocytes susceptibility to mitochondrial inhibitors. Cuprizone a copper chelator which interferes with complex IV of the mitochondria [27-29]. Cuprizone induced oligodendrocytes death followed by complete demyelination in a mouse brain [28]. When cuprizone treatment was stopped at 5 weeks, remyelination occurred within 2 weeks. However, if cuprizone was maintained for up to 16 weeks, OPCS and newly formed oligodendrocytes were destroyed [29,30].
Rotenone, like cuprizone, is known to interfere with mitochondrial oxidative phosphorylation and the production of energy. Rotenone is an inhibitor of mitochondrial NADH dehydrogenase (complex I), one of the five enzymes of the inner mitochondrial membrane involved in oxidative phosphorylation [31,32]. This inhibition prevents the transfer of electrons from the iron-sulfur center in complex I to ubiquinone which interferes with ability of NADH to create ATP, thus lowering the intracellular ATP levels. Since complex I is unable to pass off its electrons to coenzyme Q10, there is a back-up of electrons in the mitochondrial matrix. Cellular oxygen is reduced to its radical, which creates Reactive Oxygen Species (ROS) [32,33]. Rotenone is highly hydrophobic, able to easily diffuse and pass through both the blood brain barrier and plasma membrane [34]. Additional studies have shown rotenone's ability to significantly increased levels of NO [35] microglia and increase expression of both caspase-1 and mature IL-1β [36].
Because of the structural, cellular and functional differences between the mouse and the human cortices, the need for studying the human brain has become more and more compelling. In 2013, the Lancaster group published a paper presenting a novel approach towards studying human neurodevelopmental processes through in vitro culture of cerebral organoids derived from embryonic stem cells (ESC) or induced pluripotent stem cells (iPSCs). The organoids, or "mini brains," provided new, outside the uterus insights into early human brain development and disease etiology [37,38]. These cerebral organoids mimic and closely model human brain development by generating the cerebral cortex, ventral telencephalon, choroid plexus, and retinal identities, among other brain regions [39]. The production of organoids in vitro can be used for studies of cell migration, differentiation, basic early neurodevelopment, as well as disease development and progression. The cerebral organoids allowed for the first time to relate the broad Integrative Nuclear FGFR1 Signaling (NFS)-linked transcriptional dysregulations found in schizophrenia iPSC neural progenitor cells (NPC) [40] to the development of human brain malformations in schizophrenia and prenatal mother immune attack (MIA) [38,41]. Our laboratory recently showed formation of oligodendrocytes and myelination of cortical neurons in human and their disruptions by genetic factors (schizophrenia) and environmental factor, cytokine TNFα [41].
In the present study we used rotenone to model hypoxia induced inhibition of mitochondrial oxidative phosphorylation, OXPHOS. To model their effects on human brain development, we exposed developing human iPSC derived cerebral organoids to rotenone. We found that after transient oxidative insult, the depleted oligodendrocyte population is capable of significant spontaneous regeneration and that Phenylvutyrate which stimulates acetyl-CoA and energy production from glucose enhance the recovery.
Materials and Methods
Cells and organoid cultures
Induced pluripotent stem cells (iPSCs) (described in the previous studies: 38, 40), Table 1 were derived from healthy, (control) subjects. These iPSC lines were used in our previous studies producing similar RNAseq and cerebral organoids results [38,40]. In all lines the effects of rotenone were similar and thus appeared to be gender- and donors' age-independent (Table 1).
Cells were cultured and cerebral organoids were generated based on our modified protocol [38,41].
Rotenone exposures
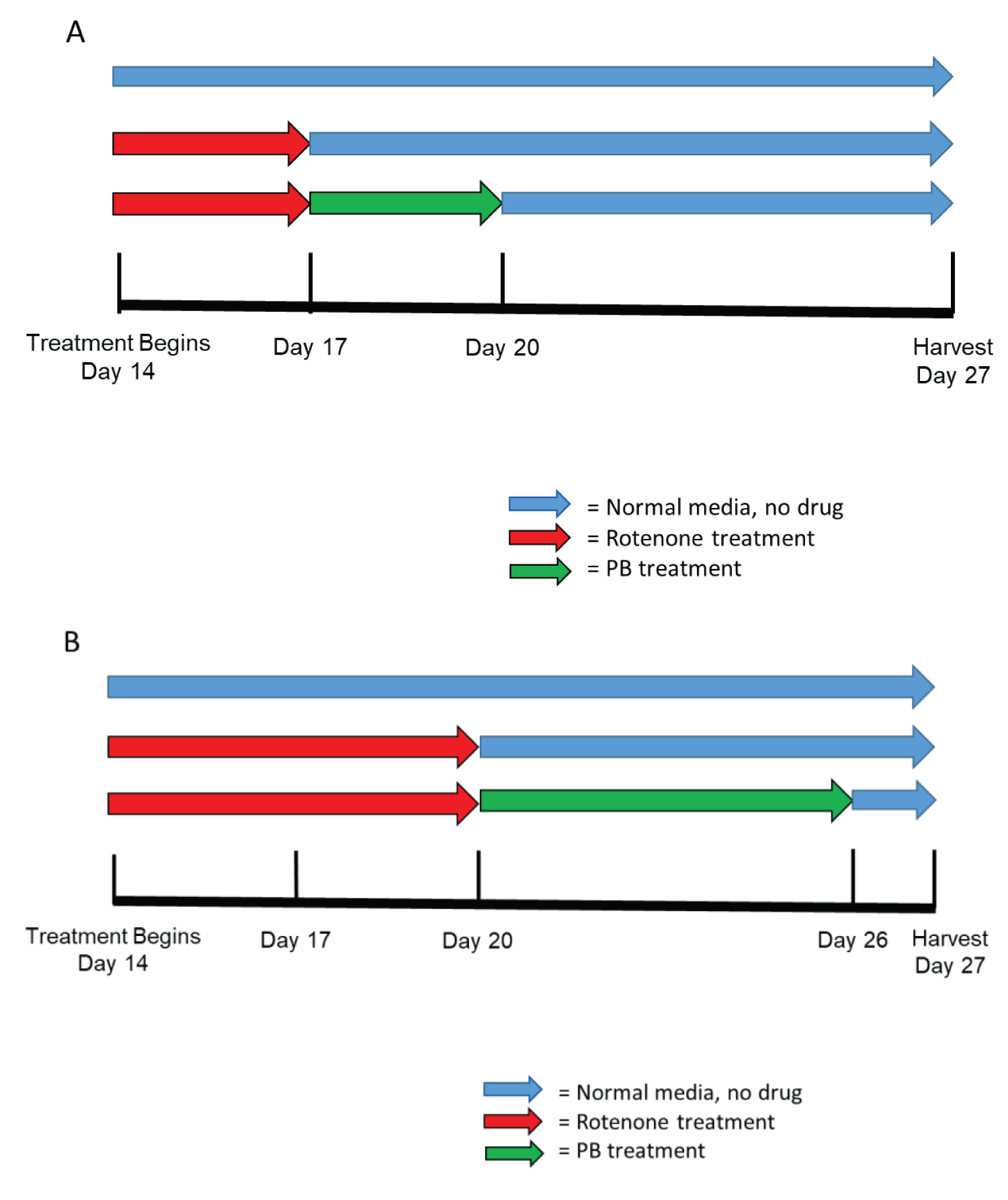
Exposure started at day 14th of cerebral organoids development, during active phase of cortical development [38]. Randomly selected organoids were incubated in medium containing rotenone (R8875, Sigma-Aldrich) at a concentration of 0.5 μM. At least 3 organoids from each of the 3 different cell lines were included per control and treatment paradigm. There were two different rotenone treatment groups. Group #1 exposed to rotenone for 3-days (day 14-17), three organoids were harvested at day 17 along with their control counterparts cultured in rotenone free media. In addition, 3 rotenone exposed organoids were allowed a 10-day recovery in rotenone free media and were harvested at day 27 along with the control counterparts. Group #2 of organoids were exposed to rotenone for 6-days (days 14-20). Three of these organoids were harvested at day 20 and 3 after an additional 7-days (day 27) culture in rotenone free media, along with their control counterparts (Figure 1). Chronic rotenone exposure was found to disrupt mitochondrial networks and to increase the ratio of mitochondria fusion to fission in cultured neurons [42]. In Supplemental Figure S1 we show disruption of the mitochondrial networks in cerebral organoids exposed to rotenone for 3-days.
Phenylbutyrate treatments
At the end of the rotenone treatment 3 organoids of group 1 were treated for three days (days 17- 20) with 1.0 μM sodium Phenylbutyrate (PB) (SML0309, Sigma-Aldrich) and harvested at day 27. Three organoids exposed to rotenone at days 14-20 in group #2, were treated with PB for 6-days, days 20-26, maintained in drug-free control media for 1 day and then harvested at day 27 (Figure 1).
Immuncytochemistry
Organoids from control and treatment were harvested at multiple time points, day 17, 20, or 27 (Figure 1). Organoids were fixed with 4% paraformaldehyde diluted in 1x PBS (Polysciences, Inc.) for 25 min at room temperature on a shaker followed by 3 washes in 1 × PBS for 10 minutes each. Organoids were cryoprotected in a gradient of sucrose solutions (Sigma Aldrich) 7.5%, 15% and 30% for cyroprotection at temperature 4 °C and transferred to 7.5% gelatin diluted in 10% sucrose. Subsequently, organoids were incubated at 37 °C for 20 minutes to assure for penetration of gelatin to superficial layers of organoids and protect against further separation from gelatin embedding medium. Once organoids in gelatin blocks the organoids were frozen in liquid nitrogen and stored in -80 °C freezer. Frozen organoids were sectioned on Cryostat into 30 μM sections and dry-mounted on an adhesive microscopic glasses (Matsunami, MAS-GP). Each organoid produced approximately 10 slides each with 4- 5 individual sections.
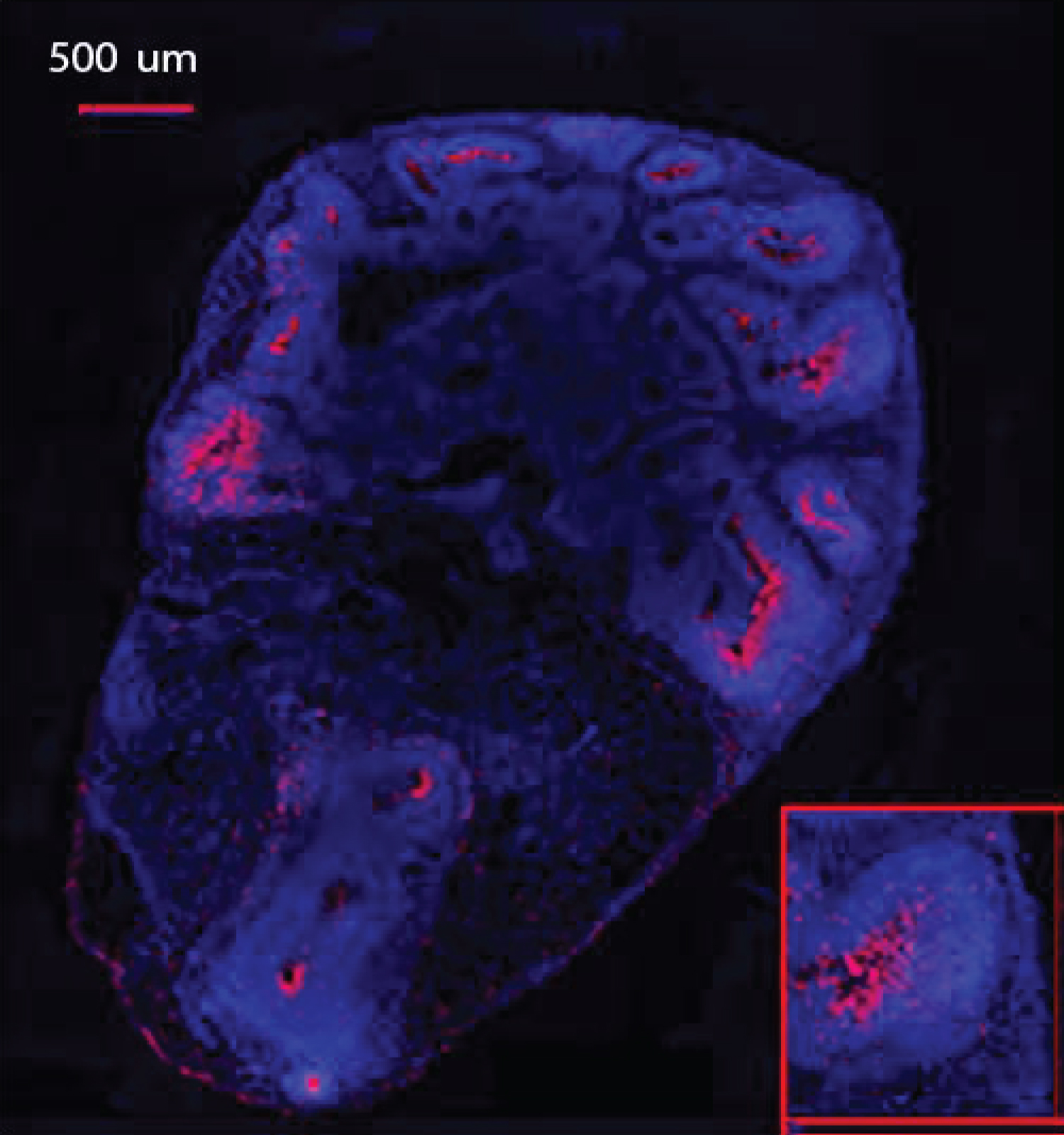
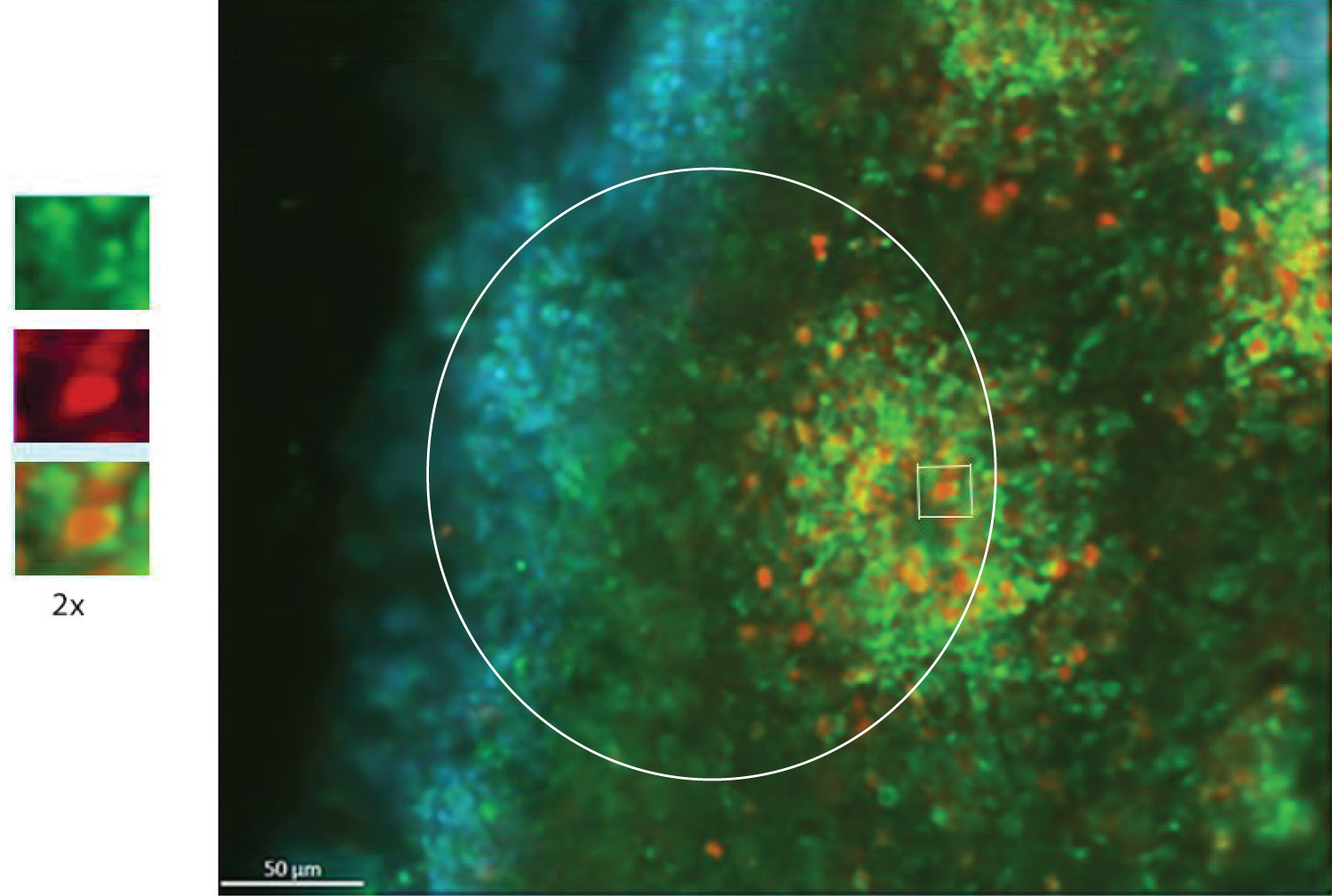
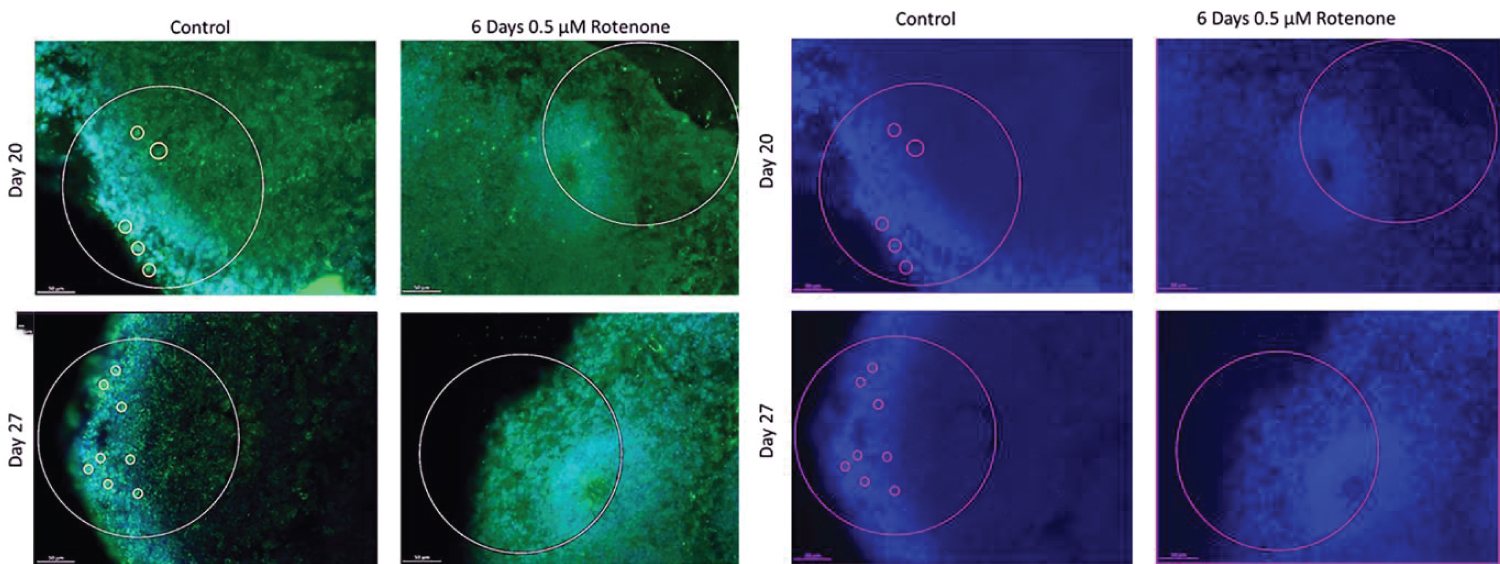
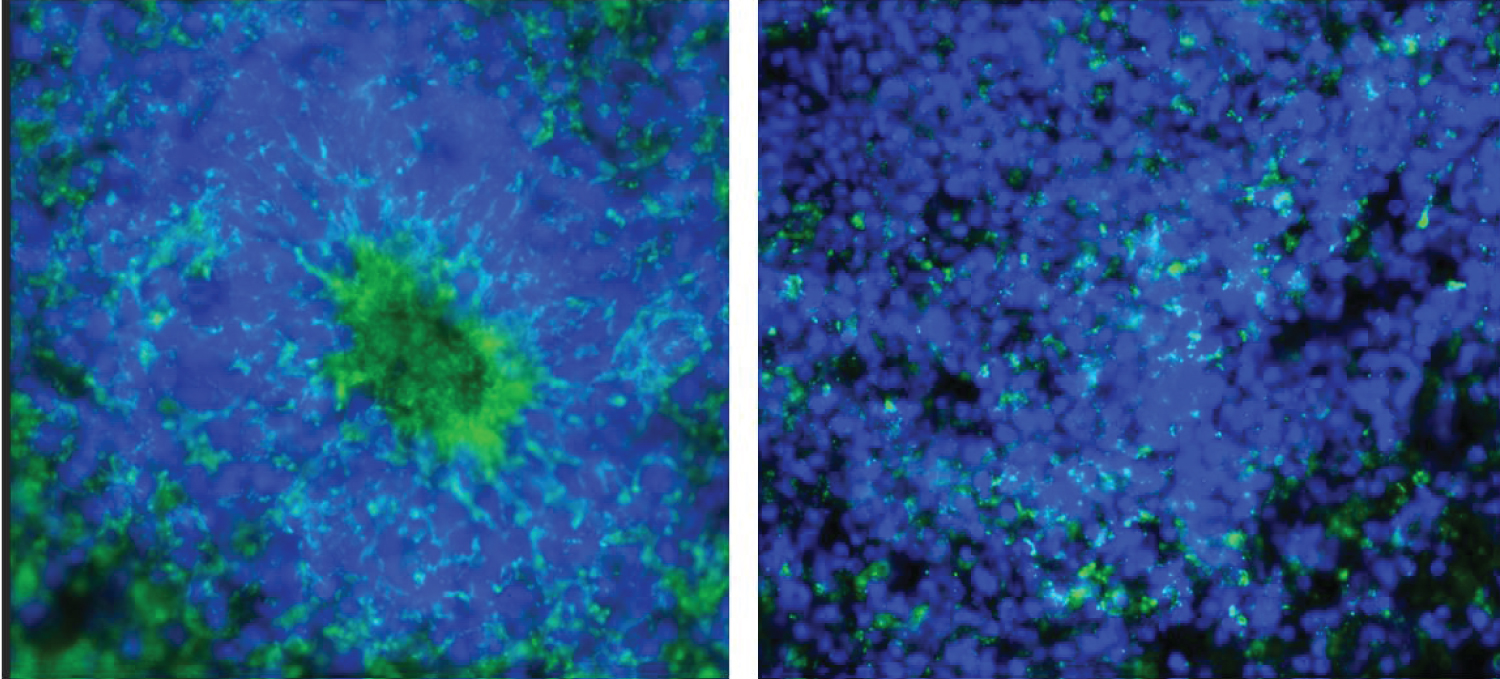
Sections were stained in batches consisting all representative sections from each experimental group and control, as described in Benson, et al. [41]. The following primary antibodies were used: Anti-Ki67 polyclonal (rabbit) antibody (Cat #: ab15580, abcam) proliferation marker, and mouse monoclonal anti-O4 antibody IgM at dilution 1:330 (Cat #: MO15002.9, Neuromics), oligodendrocyte Marker. The following secondary antibodies were used: Alexa Fluor® 568 goat anti-rabbit IgG (H+L) polyclonal Antibody at a dilution of 1:1000 (Cat. # A11011, Life Technologies - Molecular Probes) and Alexa Fluor® Goat pAb to Mouse IgM (Cat # ab150121, abcam). Figure 2 shows example section double immunostained for Ki67+ (red) and O4+ (green) and with the nuclei stained with DAPI (blue).
Microscopy, image and statistical analyses
Zeiss Axio Imager Upright Fluorescence microscope was used, equipped with Zen Blue 2.3 software. Images were taken at 5 × to create a super image and at 20 × magnification in order to capture each individual rosette in the organoid structure.
All images were opened in Zen Blue and the red Ki67 channel was isolated. Each ROI included, the center of the rosette, ventricular zone (VZ), intermediate zone (IZ), and the cortical zone (CZ) region. ROI delineated with a white circle of an area 54,609.718 μm2. All images were analyzed for Ki67+ cells using a MATLAB script written by Siddhartha Dhiman in Stachowiak, et al. [38]. The MATLAB program runs two simultaneously processes together. Through strict cellular dimensions the program identifies each Ki67+ cell and the shortest distance between each of the cells, called minimum spanning tree (MST) analysis. MST shows the least distance between cells in a ROI reflecting cell separation.
O4+ cells were counted manually from the same images that were used for Ki67 analysis. In order to quantify cells that were double stained for Ki67 and O4, the original O4 image was pulled up next to the KI67 image showing the individual Ki67+ cells identified by MATLAB script. All statistical analyses were performed using IBM SPSS software provided by the University at Buffalo. Figures show a representative images with the white circles including the center of the rosette out to the cortical region.
Statistical analyses
The effects of individual Rotenone treatment were analyzed using 1-way Analysis of Variance (ANOVA). Rotenone plus PB were analyzed using 2-way ANOVA. Significant ANOVA scores were followed by post-hoc comparisons between multiple samples using Least Significant Difference (LSD) test. Mean + Standard errors were employed for all graphical representations. Analysis was performed on images from 3 different organoids from each of the 3 different cell lines mentioned in Table 1. This provided an N = 9 for each condition analyzed.
Results
Effects of rotenone treatments
Ki67+ cells count & distances between cells: Cerebral organoids develop distinct structures: Ventricular zone, intermediate zone, cortical zone, and marginal zone reminiscent of the human brain structures developing in the 1st and early 2nd trimesters. As previously shown [38,41], most of the Ki67+ cells were located in rosettes in the ventricular zone, where the proliferating stem/progenitor cells reside. Few of the Ki67+ cells migrated to the intermediate and cortical zones. These Ki67+ cells represent NPC [38,39,41]. The NPC are known to differentiate to neuronal, astrocytic and oligodendrocytic lines [43].
Independent of the rotenone exposure duration (3- or 6- days), rotenone induced an overall statistically significant reduction in the number of Ki67+ cells, relative to the control non-treated organoids (p < 0.001). When broken up by treatment periods, this statistical significance was maintained.
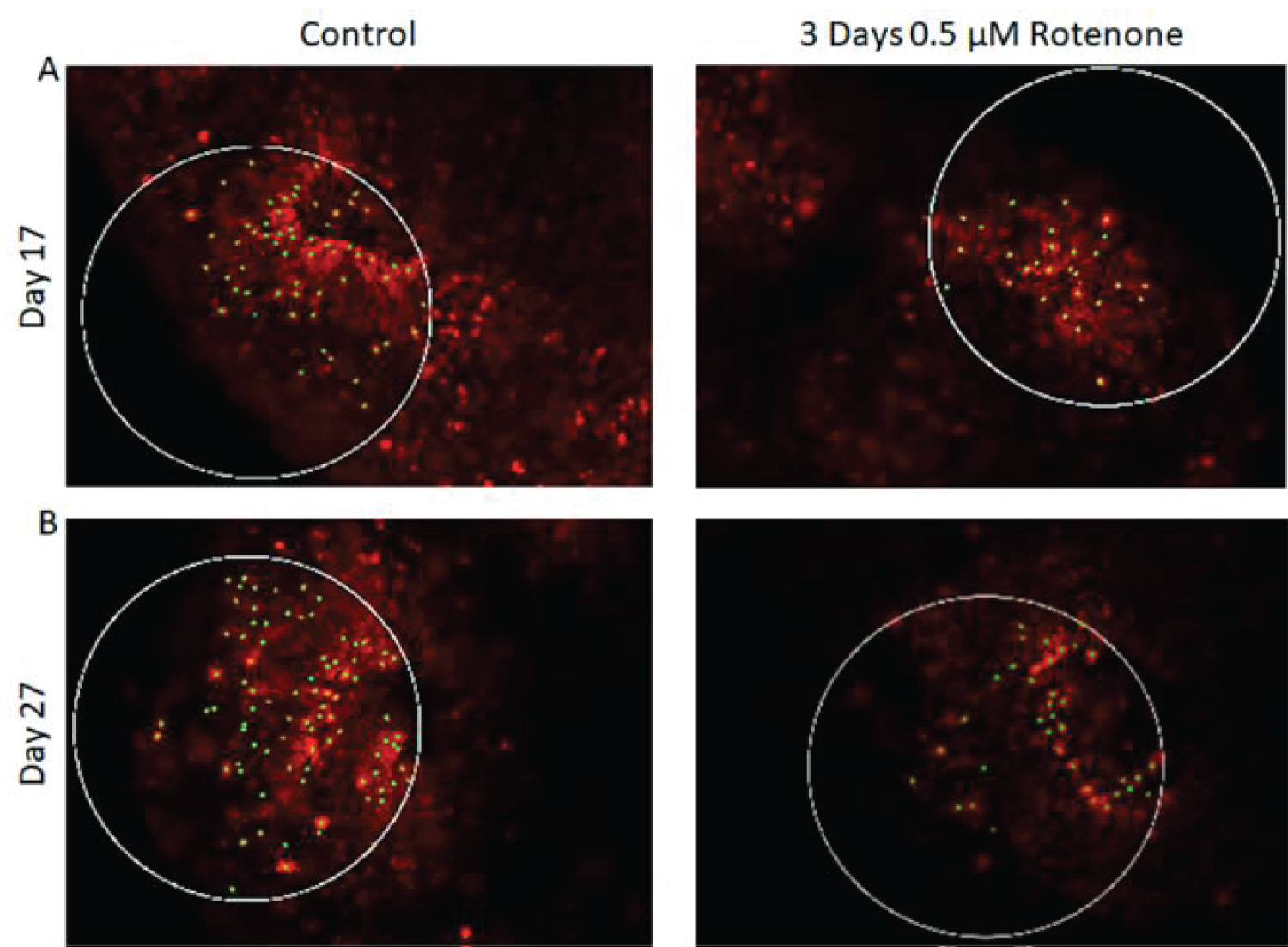
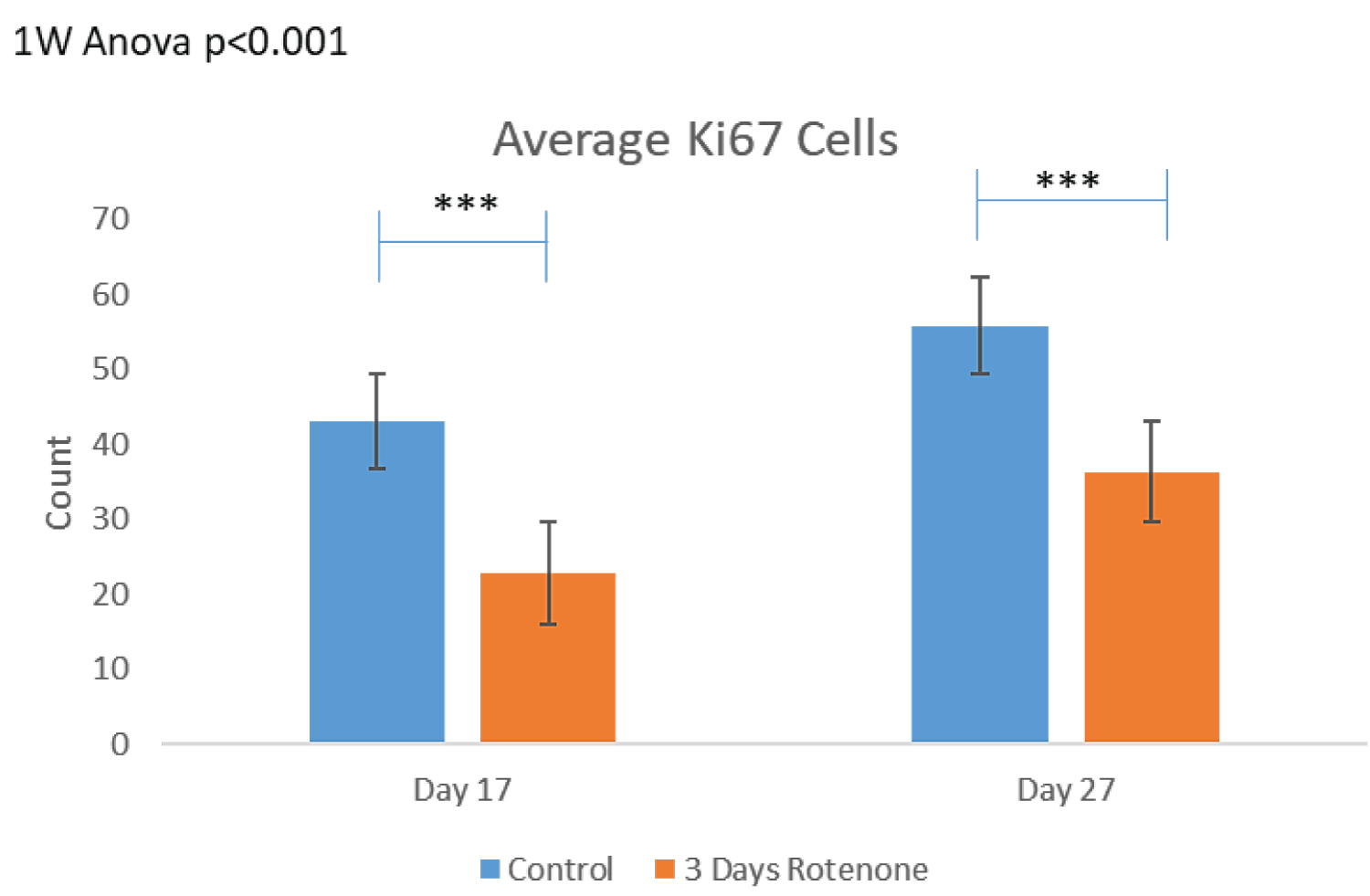
The results of 3-days Rotenone exposure are shown on Figure 3. Figure 3A shows representative images of Ki67 stained sections with outlined circular ROI. Images taken from the output folder from the MATLAB program show that at the end of 3-days (days 14-17) of Rotenone exposure, the organoids had reduced number of Ki67+ cells compared to control (Figure 3A). When Rotenone treatment was followed by drug free 10-day recovery period, at day 27 the number of Ki67+ cells remained markedly depleted (Figure 3A).
Figure 3B shows the results of the computer-based cell identification and automated counting.
There was a statistically significant decrease (p < 0.001) in the density of Ki67+ cells per ROI following 3-days of Rotenone exposure from 43.12 ± 4.81 in control to 22.82 ± 2.75. The organoids cultured in control drug-free conditions continued to grow from days 17 to 27, where the average number of their Ki67+ cells per ROI increased to 55.83 ± 6.35, a trend that was not statistically significant. When the rotenone exposure was followed by a 10-day rotenone-free recovery period the average number of Ki67+ cells increased significantly (p < 0.05) to 36.33 ± 2.61, though it was still significantly reduced compared to organoids raised in rotenone-free control condition (p < 0.001).
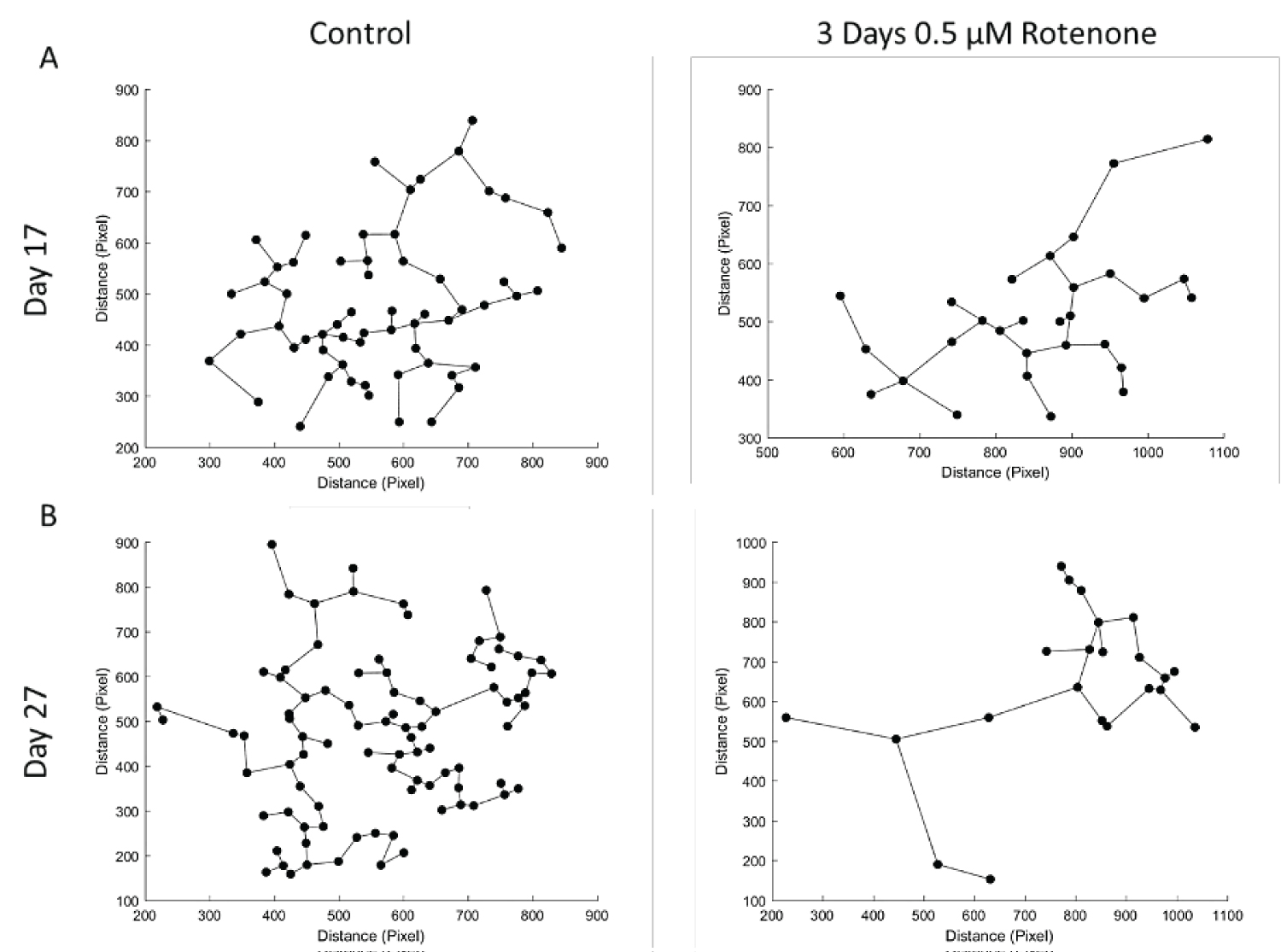
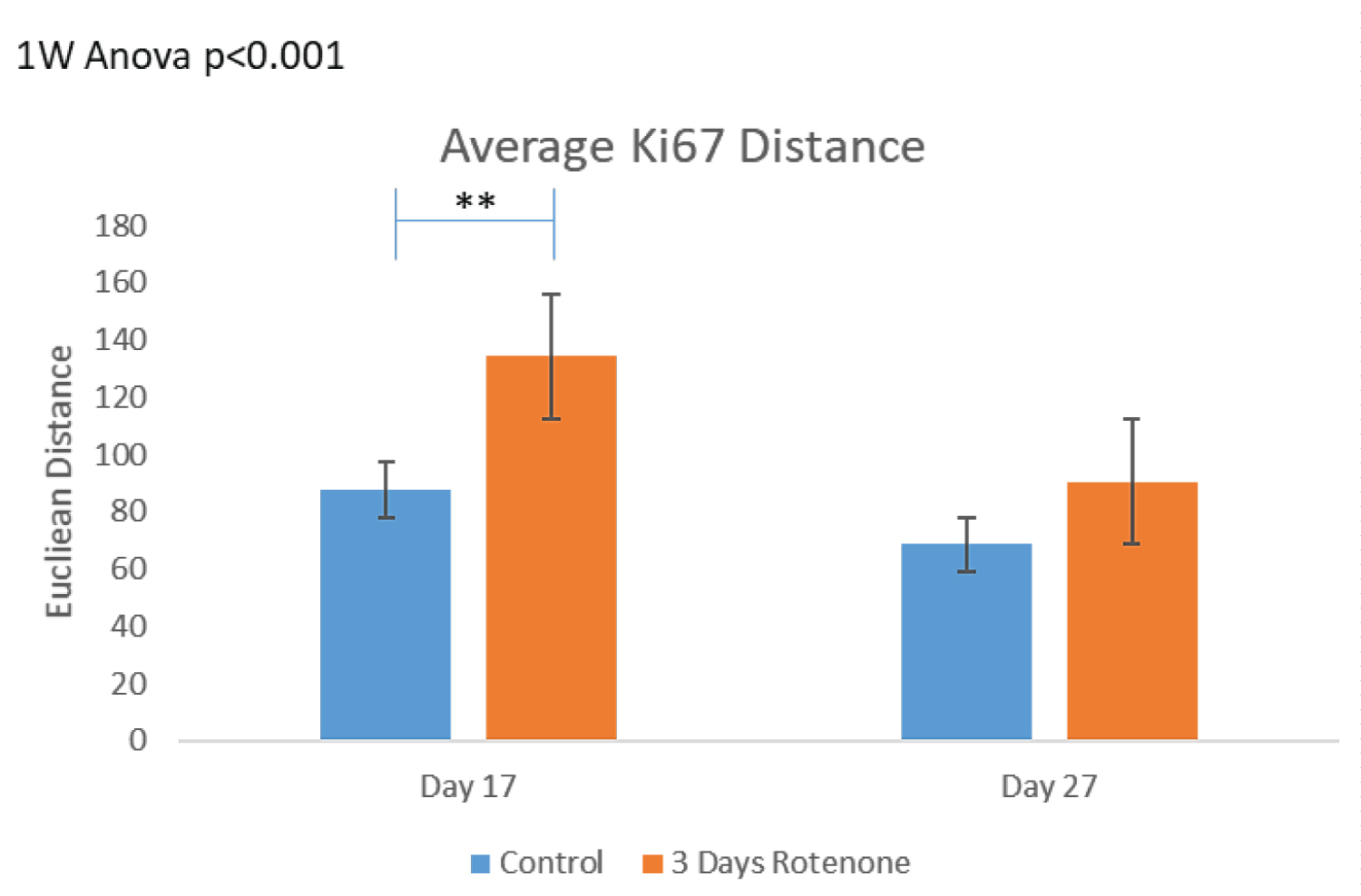
To assess distances between proliferating (Ki67+) cells, we performed MST analyses as described in Stachowiak, et al. [38]. Figure 3C shows the MST analysis performed by the MATLAB program on the images from Figure 3A. Figure 3D shows results of MST measurements in all ROI analyzed. In the control conditions, cells were grouped close to each other and following 3-days of Rotenone treatment the average MST distance between cells increased significantly compared to control (p < 0.01) at 17 days.
However, at day 27, following an additional 10-days without rotenone, the average distance between cells was no longer significantly different in rotenone exposed organoids from the control levels (p = 0.239) suggesting that these differences may not be due to the cells loss, but potentially reflect changes in cell migration.
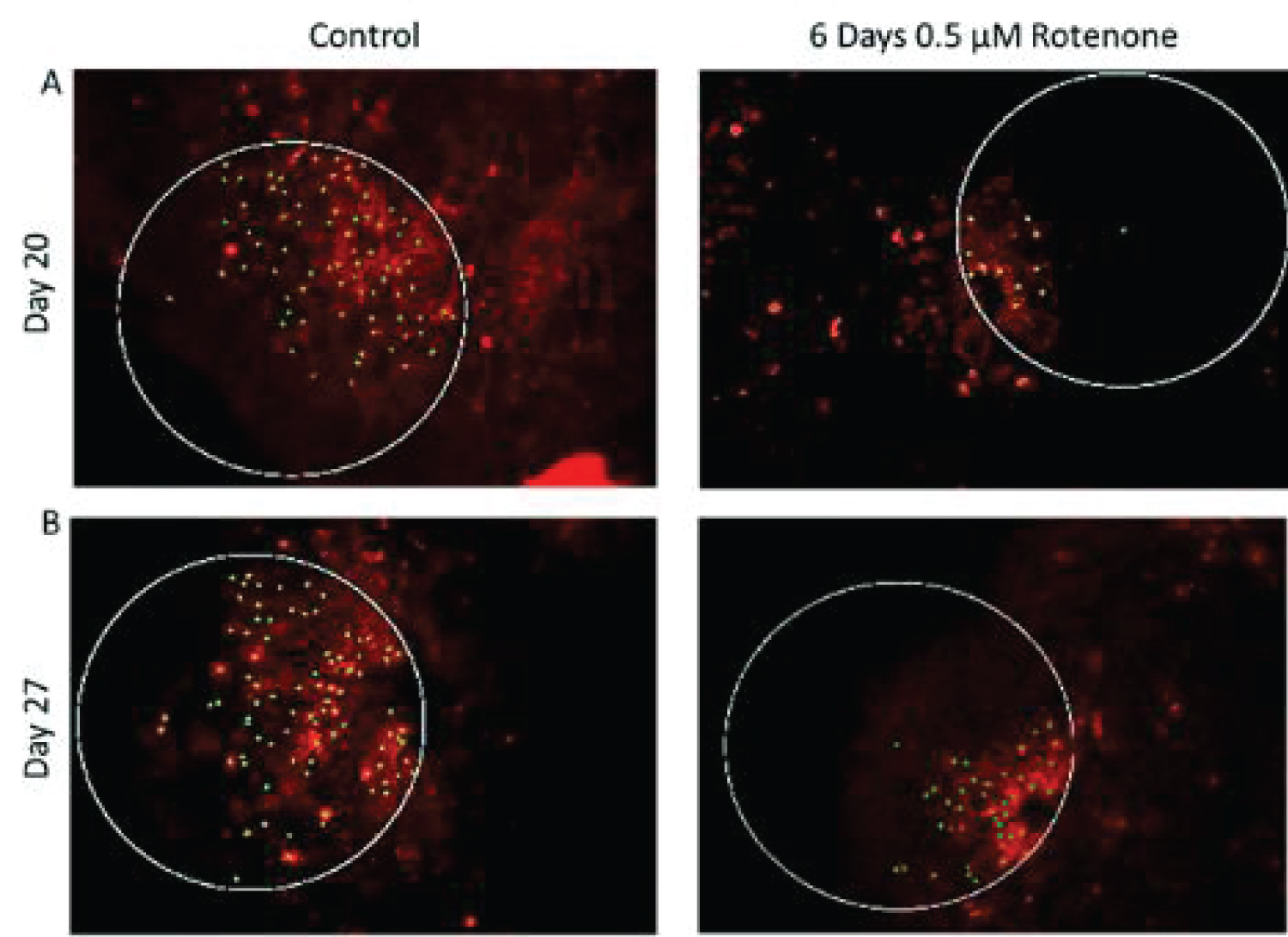
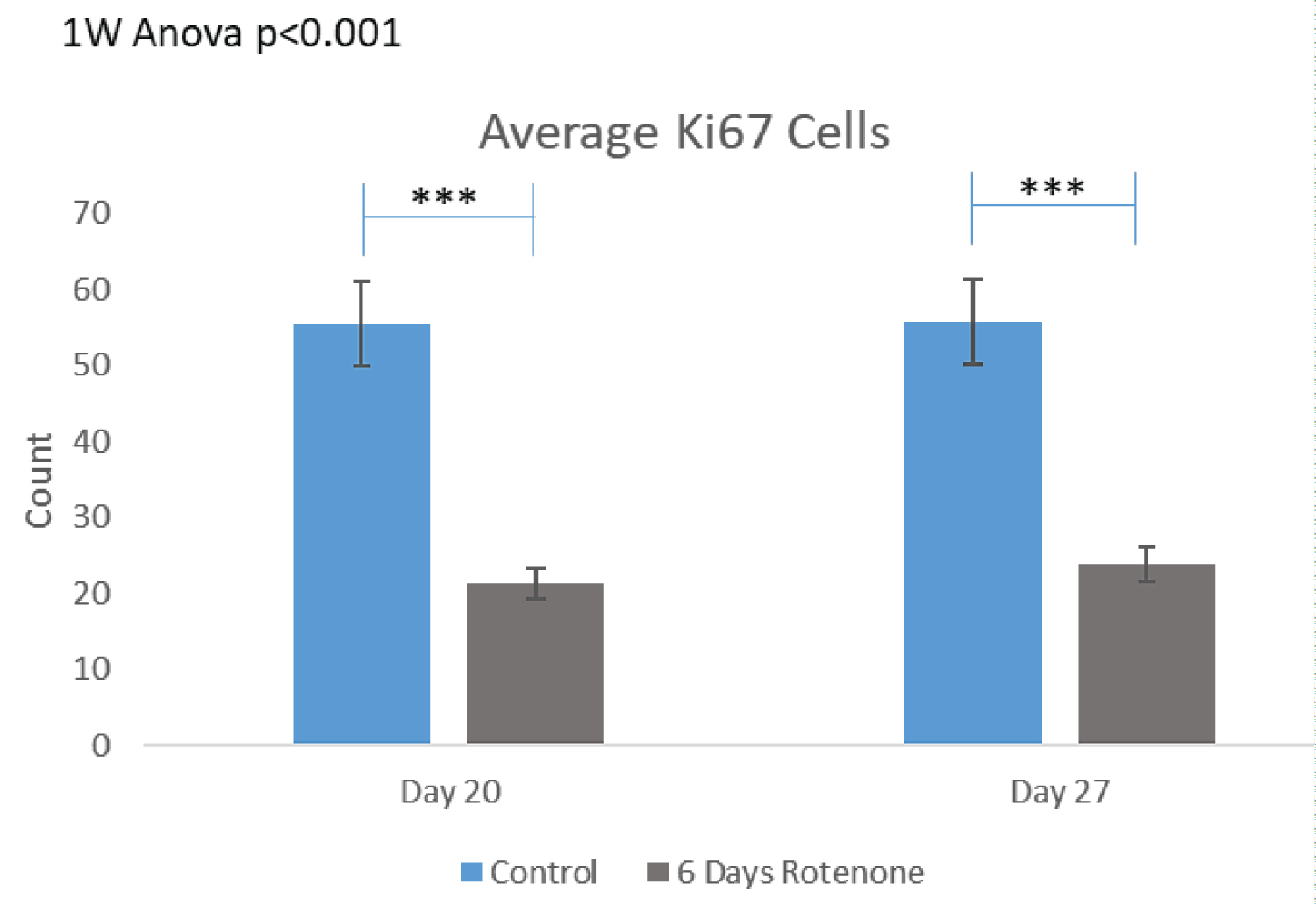
The results of 6-day rotenone exposure during days 14-20 are shown on Figure 4. At the end of Rotenone exposure (day 20), the organoid had fewer Ki67+ cells (Figure 4A and Figure 4B) compared to control. This amounted to two-fold reduction (from 55.83 ± 5.53 in control to 21.34 ± 3.01 in 6-days of rotenone (Figure 4B) which was a statistically significant (p < 0.001). The rotenone induced depletion of the Ki67+ cell density remained significant at day 27, following 7-day rotenone free recovery at p < 0.001 compared to control but was similar as at day 20, without 7-day post rotenone recovery. Thus, the extended rotenone exposure prevented further development of proliferating NPC.
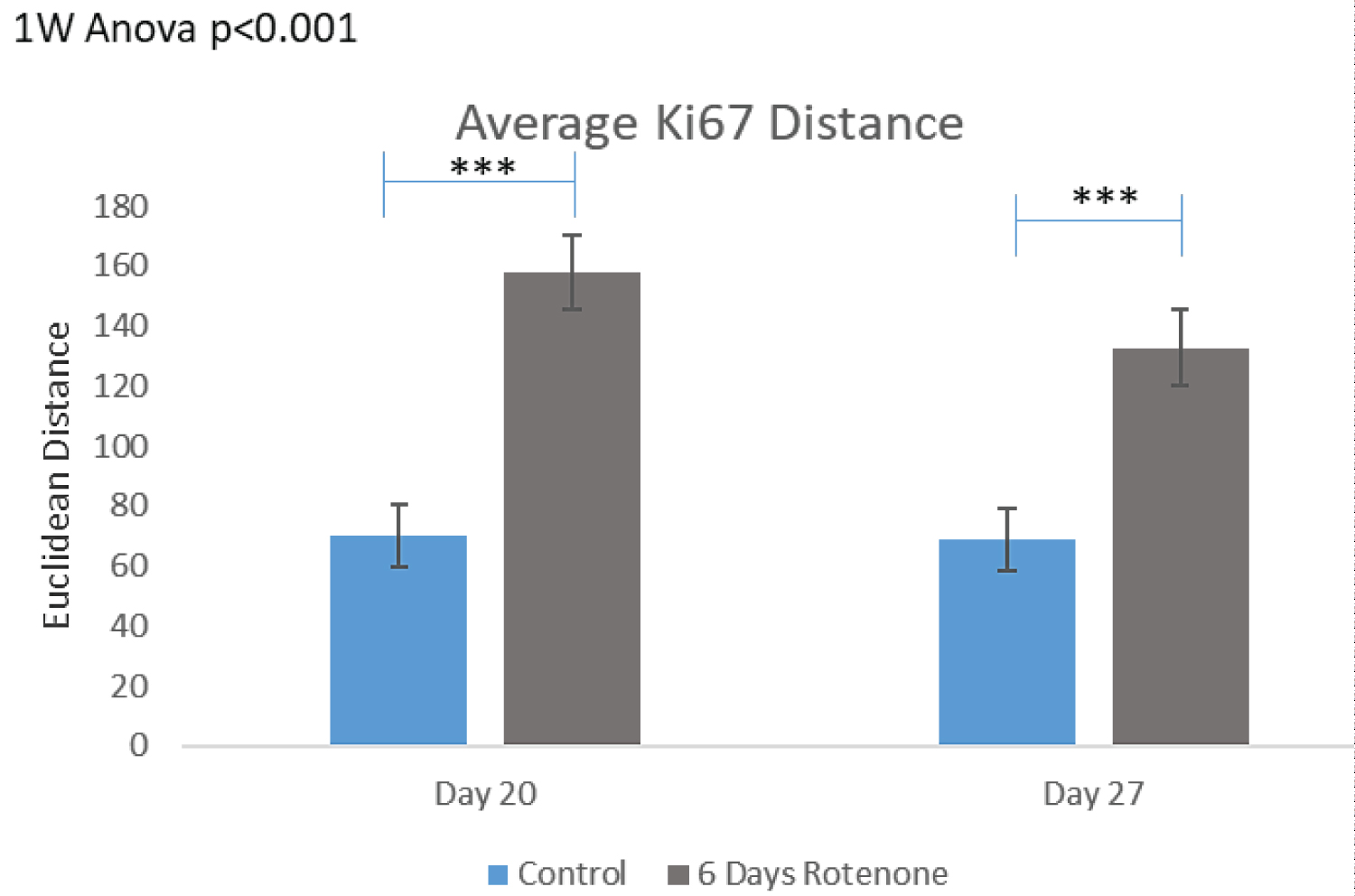
Figure 4C shows that the MST distances between Ki67+ cells were significantly increased after 6 days of rotenone (p < 0.001) and remained significantly increased following 7-days of rotenone-free recovery (p < 0.001) (Figure 4C).
Oligodendrocyte population: To determine how the rotenone may affect oligodendrocytic cells, organoid sections were stained with antibodies against the O4 protein expressed by mature oligodendrocytes and by proliferating immature Oligodendrocyte Precursor Cells (OPC). The same images and ROIs used in the Ki67+ count were analyzed. As with Ki67+ count, Rotenone caused an overall statistically significant reduction in the number of O4+ cells compared to control organoids that were not exposed to rotenone (p < 0.001). When broken up by treatment periods, this reduction remained statistically significant.
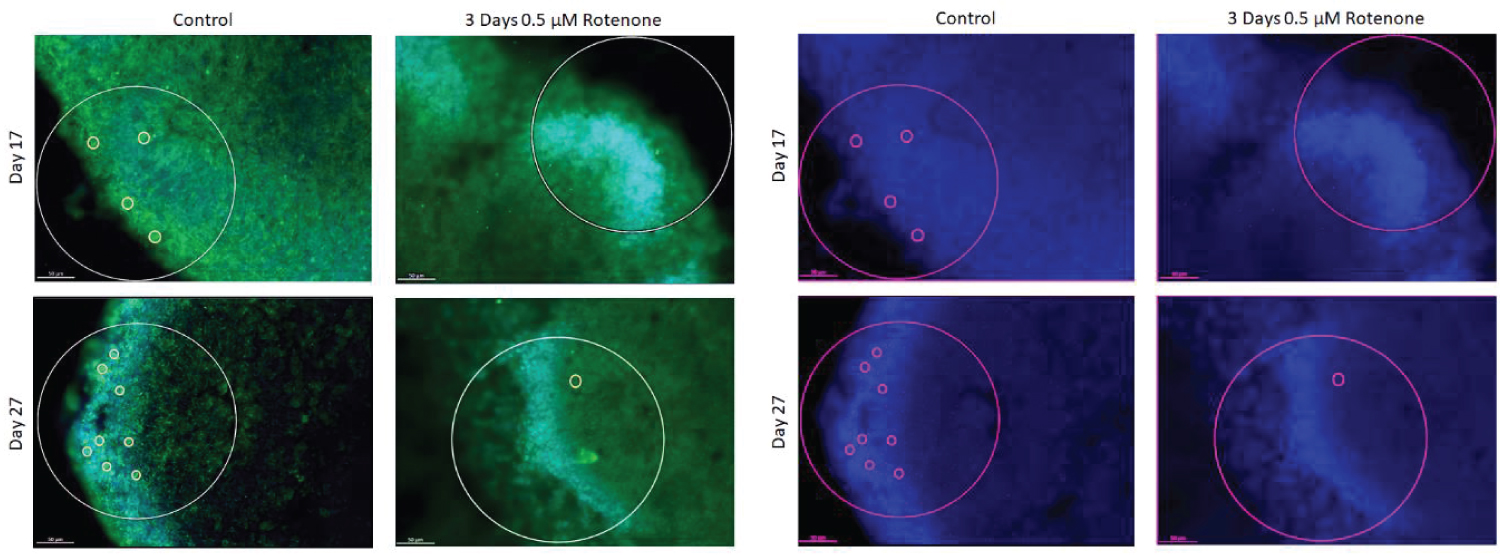
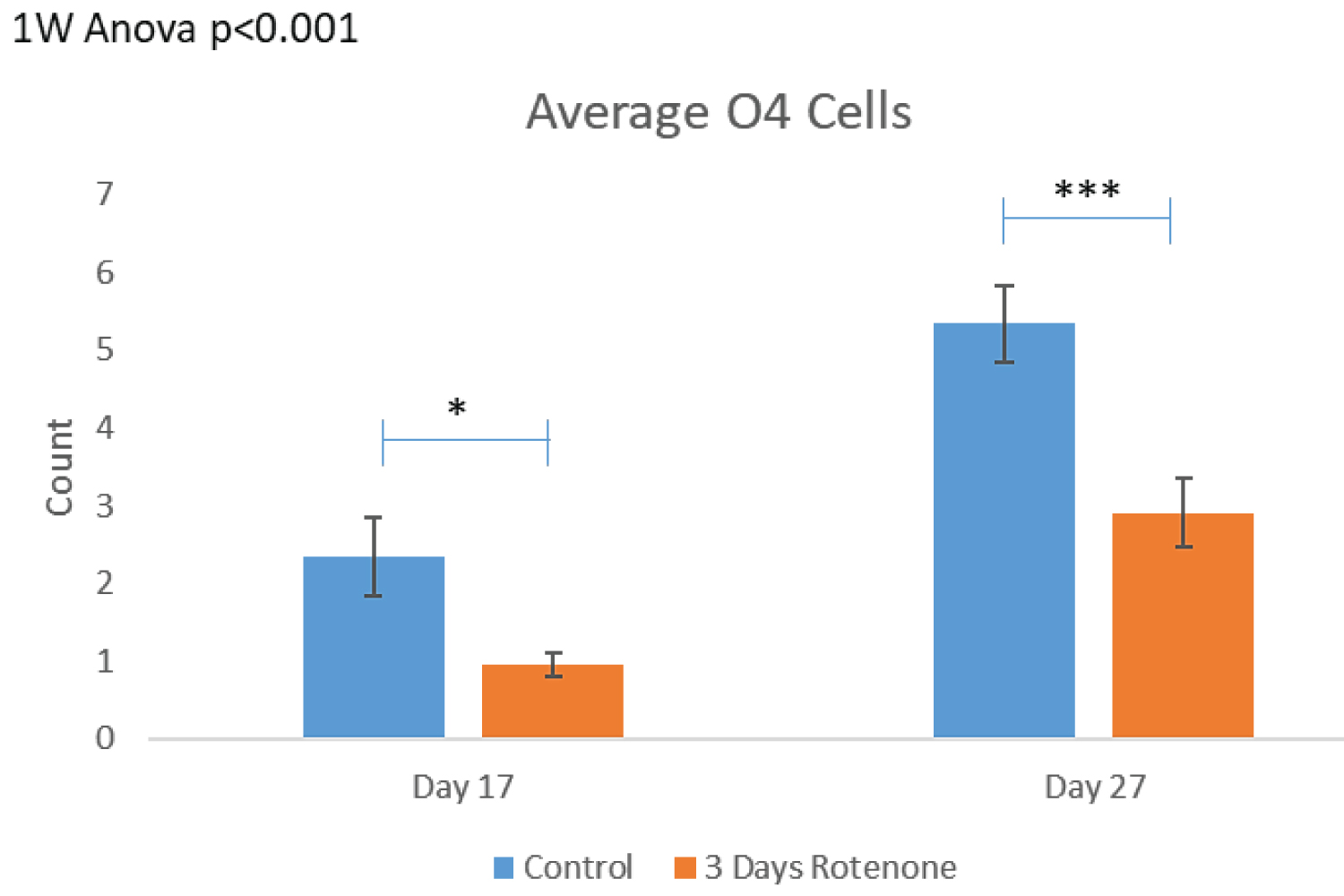
The effects of 3-day rotenone are shown on Figure 5. Figure 5A shows representative images of the organoid sections that were double stained for Ki67 and O4. The images are the same as in Figure 3A with the KI67 red channel turned off and show cells with the cytoplasmic O4 stain. There were 2-4 O4+ cells (marked by yellow circles) in the ROIs examined of control organoids. As the control organoids continued to grow and develop, number of O4+ cells visibly increased (Figure 5A) and the average numbers per ROI in control organoids increased over two-fold from 2.35 ± 0.36 at day 17 to 5.34 ± 1.0 at day 27 (Figure 5B). In contrast, in organoids exposed to rotenone for 3-days there were single or no O4+ oligodendrocytes in ROIs at the end of rotenone exposure (day 20) (Figures 5A). The average number of O4+ per ROI was reduced to 0.95 ± 0.27, a statistically significant decrease of 60% (p < 0.05) (Figure 5B).
After an additional 10-days in medium without rotenone, there was increase in the number of O4+ cells counted (Figure 5B) up to 2.90 ± 0.50 per ROI; which was statistically significant compared to day 17 (p < 0.001, Figure 5B), but remained significantly lower than in control organoids at day 27 (p < 0.001).
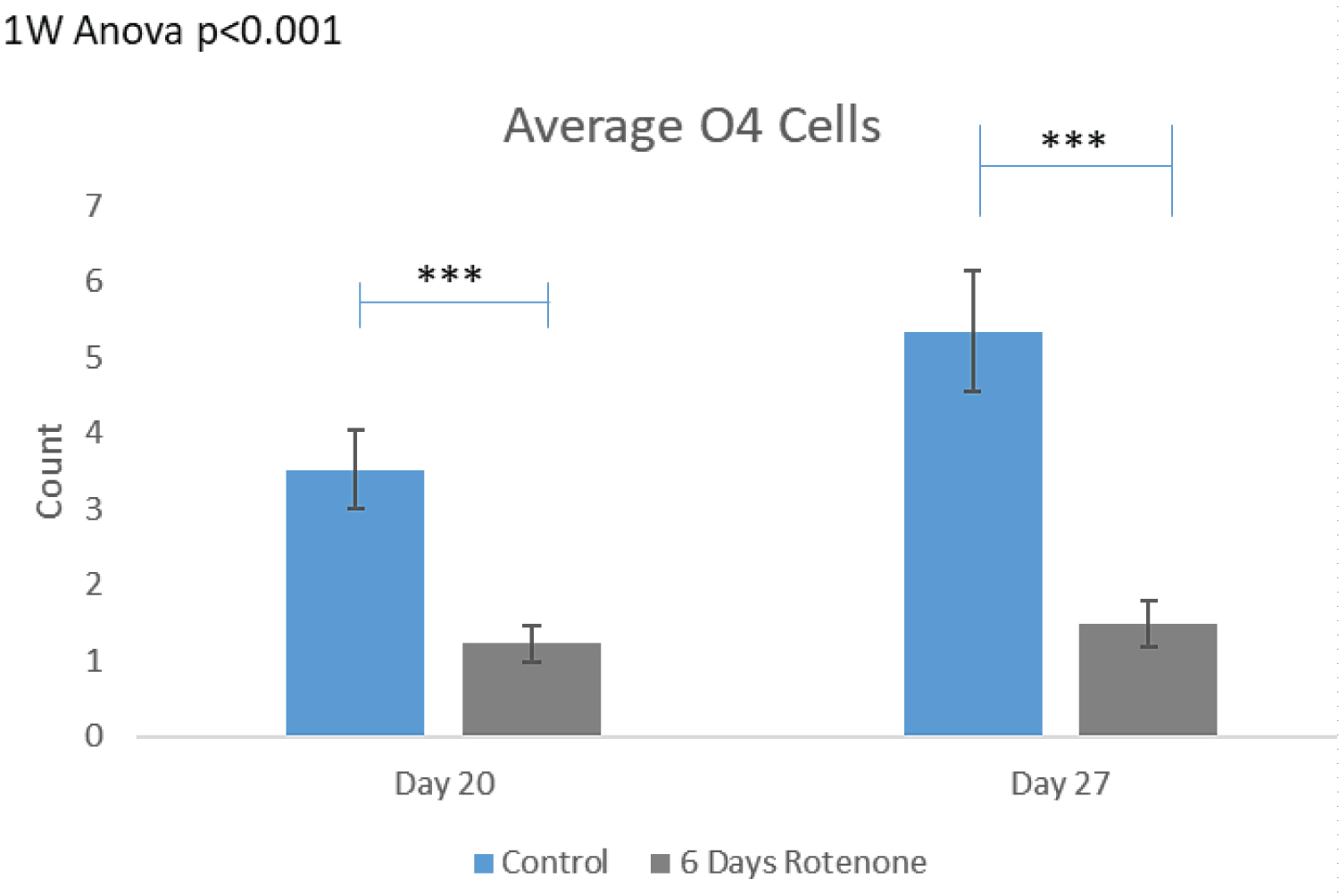
The effects of 6-day rotenone exposure are shown on Figure 6. The exemplary images on Figure 6A show no O4+ in the ROIs following 6-days of Rotenone, at day 20 or 27. In both groups, nearly 50% of the images quantified did not have a single O4+ cell counted. In Figure 6B, after 6-days Rotenone treatment (days 14-20) at day 20, the average number of O4+ cells per ROI, 1.23 ± 0.30, was significantly lower than in control, 3.53 ± 0.60 (p < 0.001). The average number of O4+ cells in control organoids at day 20 and at day 27 were not significantly different (compare Figure 6B and Figure 5B). When 6-day rotenone treatment was followed by 7-day drug free recovery period, the average number of O4+ cells remained reduced compared to control organoids (p < 0.001) but was not significantly different from the organoids harvested at the end of 6 days of rotenone exposure at day 20 (Figure 6B).
Thus, as with Ki67+ proliferating NPC, the 6-day rotenone exposure prevented further development of NPC within the time frame of our experiment, despite that in control organoids the O4+ numbers increased significantly between days 20 and 27 (p < 0.05).
OPC population: To identify the O4+ cells which represented still proliferating immature OPCs, the images of organoid sections double-stained with the Ki67 and O4 antibodies were analyzed. In control organoids, the OPC represented 57% of all O4+ cells at day 17, 47% at day 20 and 33% at day 27. On the other hand, the OPC represented only 2.5-3.3% of all proliferating Ki67+ cells.
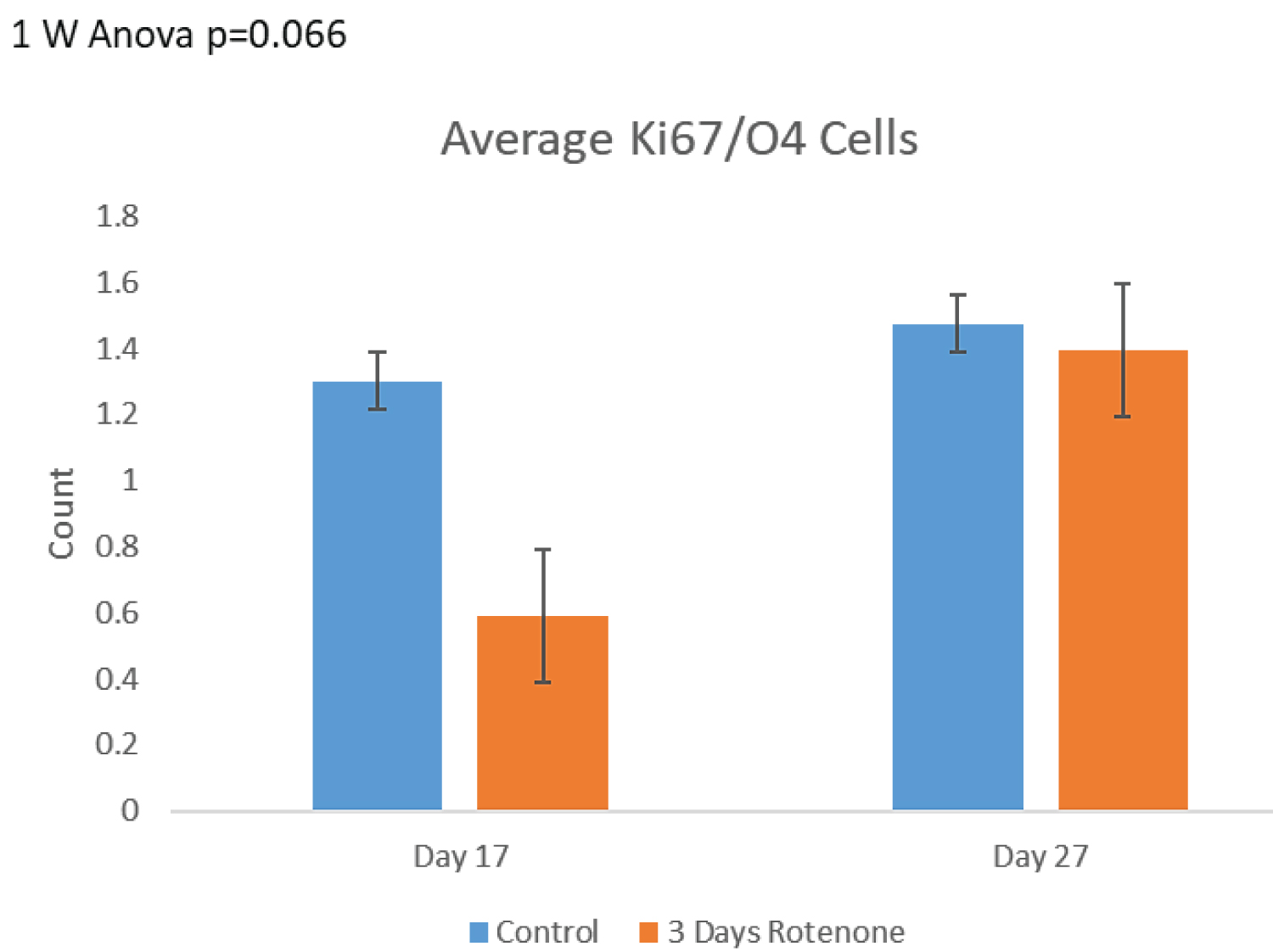
Effects of 3-days of rotenone exposure on OPC are shown on Figure 7. Following 3 days of rotenone exposure we observed a decreasing trend in the numbers of double stained O4+/Ki67+ OPC. At day 17, the number of OPC in rotenone exposed organoids was approximately 53% lower than in control organoids, similar as the total number of O4+ cells (54% loss). However, due to the small number of double stained OPC cells, the change in OPC did not attain significant levels. Following 10-day rotenone free incubation at day 27, the number of OPC showed an increasing trend towards control levels (Figure 7A). The observed changes did not reach the statistical significance (p = 0.066).
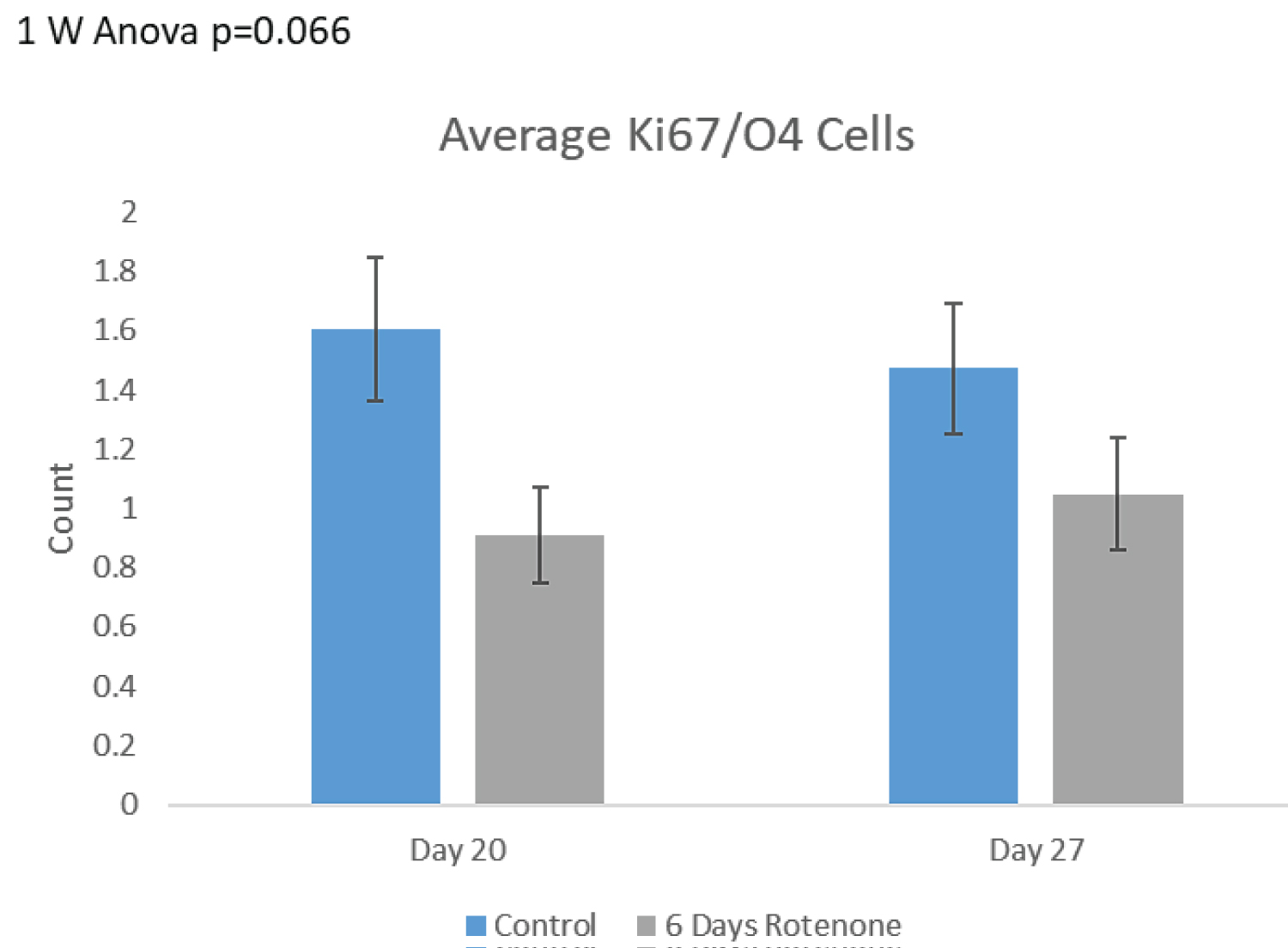
The effects of 6-days of rotenone exposure on OPC are shown on Figure 7B. We observed a decreasing trend in the OPC numbers following 6-days of Rotenone. At day 20, the number of double positive cells was approximately 50% lower when compared to control organoids and remained diminished at day 27 (Figure 7B). Thus, when provided with a 7-day post-rotenone recovery period, the number of double positive cells remained diminished and showed no recovery.
Effect of phenylbutyrate on rotenone-induced developmental changes in cerebral organoids
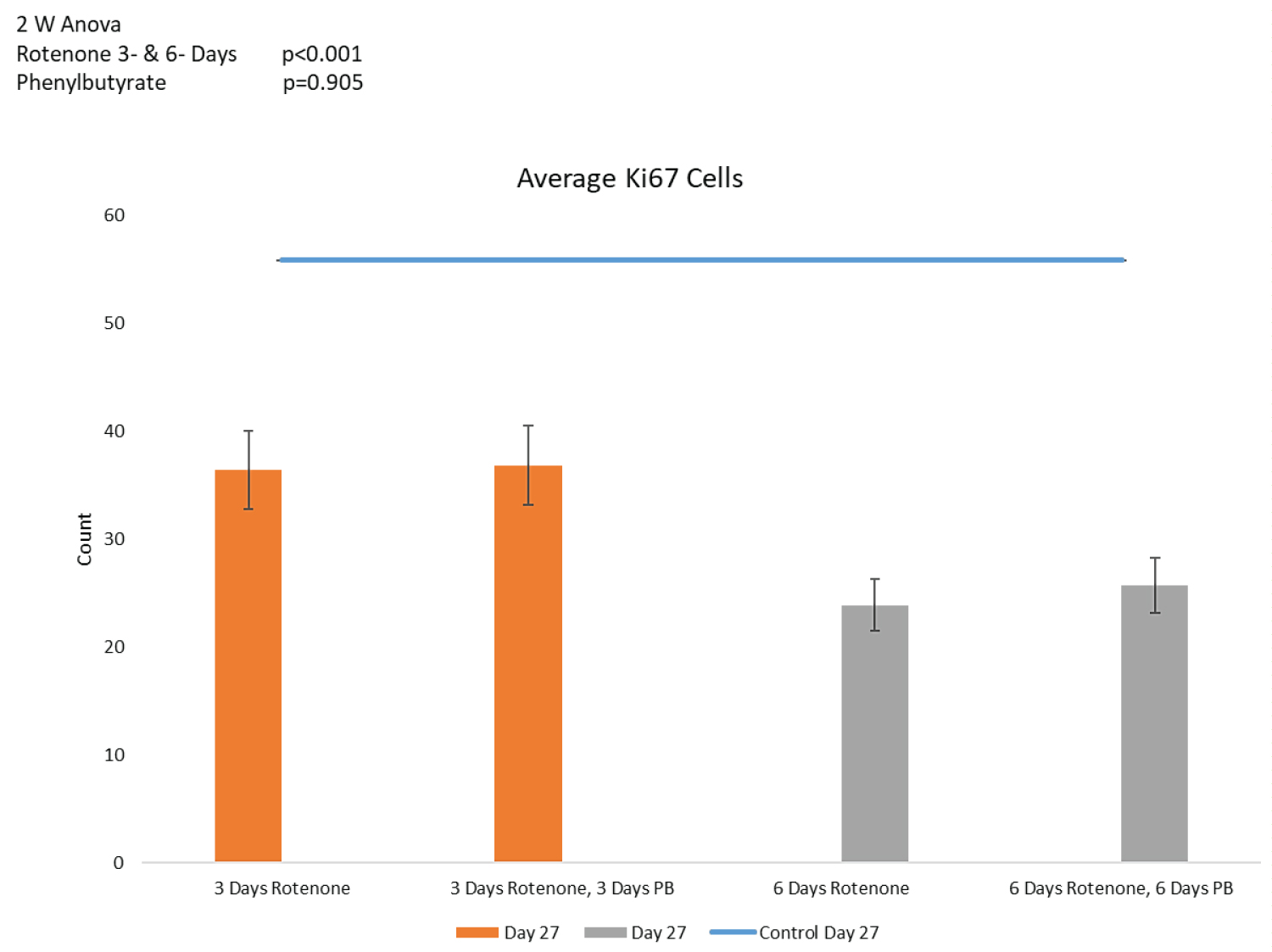
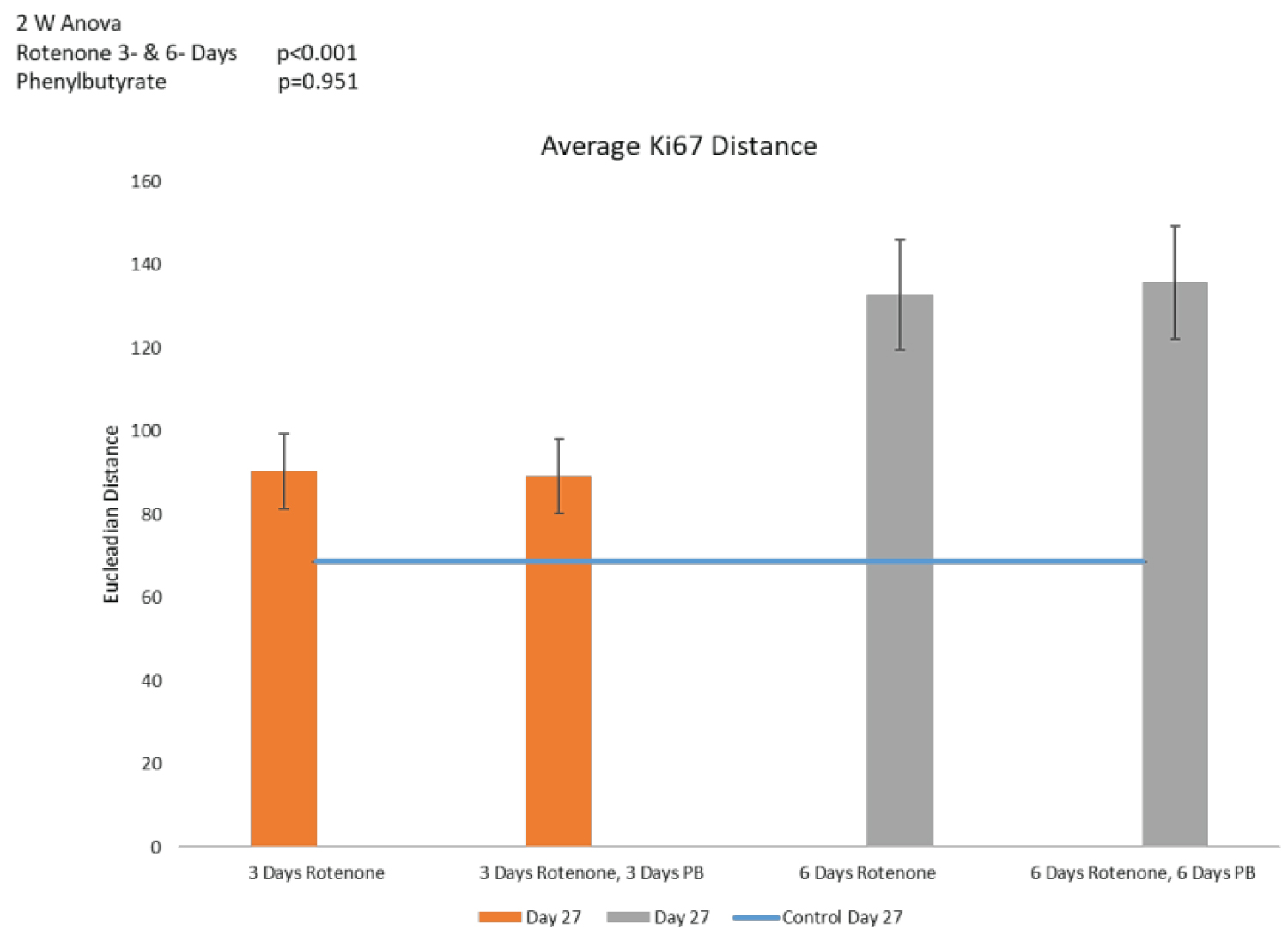
Counts of Ki67+ cells and distances between cells are shown on Figure 8A and Figure 8B, respectively. Three days of PB treatment following 3-days of rotenone (36 ROIs) had no additional effect on the number of Ki67+ cells when comparing to the rotenone exposure without the subsequent PB (Figure 8A). Similarly, following 6-days of rotenone, the 6-days of the PB treatment (42 ROIs) failed to increase the Ki67+ cell population (Figure 8A). Also, the increased distances between cells in rotenone exposed organoids were not affected by the treatments with PB as illustrated in Figure 8A. Regardless of the PB treatment the distances between cells were not significantly changed (p = 0.951, Figure 8B). Thus, treatment with PB did not reverse the rotenone-induced depletion of the proliferating cells and their migration.
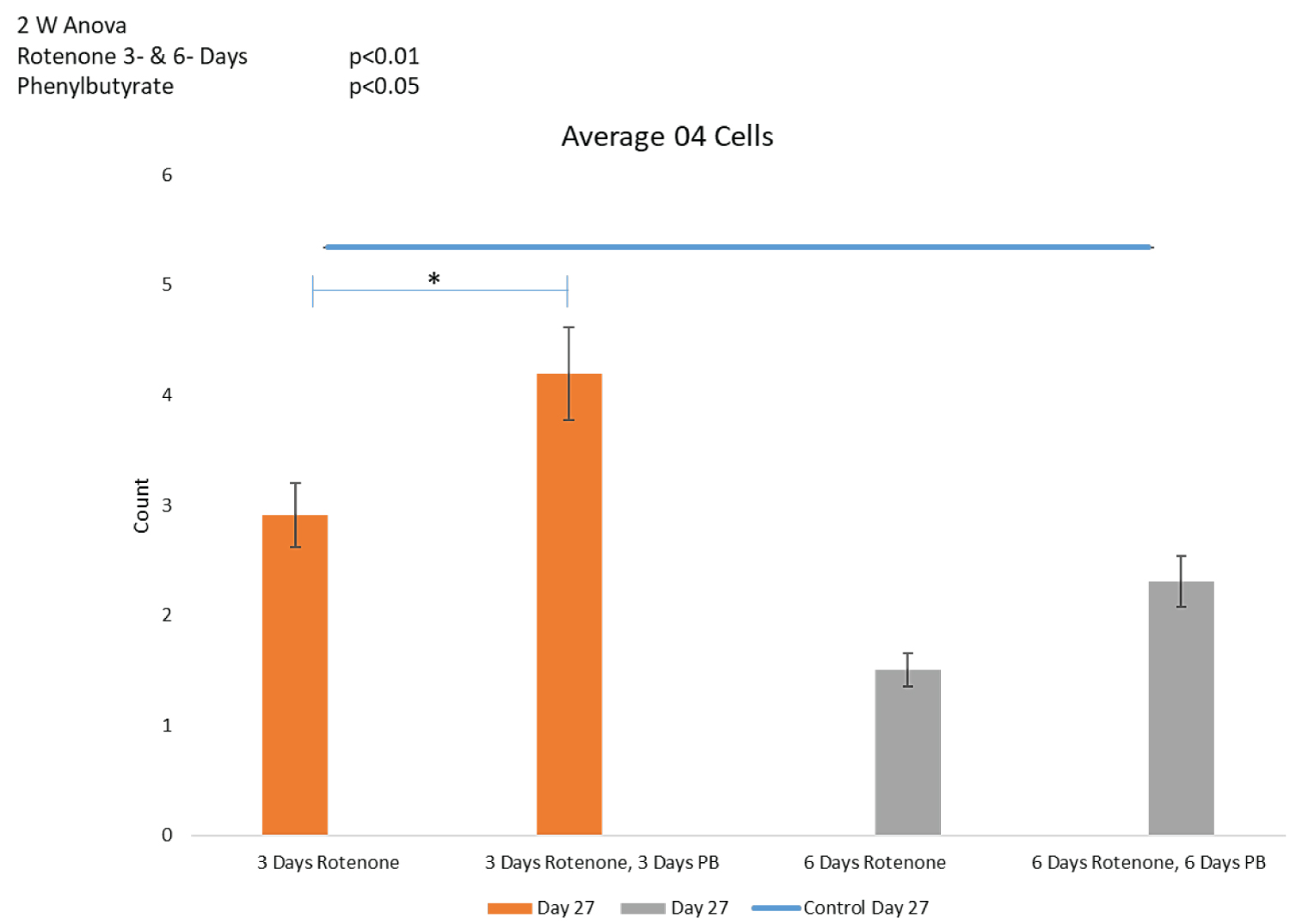
Oligodendrocyte population: 2-way ANOVA showed an overall statistically significant effect of PB (p < 0.05) on the oligodendrocyte cell population depleted by rotenone (Figure 8C). Post-hoc LSD showed that 3-days' administration with PB increased the O4+ population depleted by 3-days of Rotenone, in a statistically significant manner (p < 0.05) (Figure 8C). Thus, in addition to the spontaneous partial recovery of O4+ population during the post-rotenone 10 days incubation, inclusion of PB (3 days) was able to further significantly increase the numbers of O4+ cells at day 27 to 4.19 ± 0.6, which no longer differed significantly from the nontreated control organoids, 5.34 ± 1.0 (p = 0.112).
Figure 8C shows that 6-days of PB tended to increase the O4 population, though this increase approached but did not reach statistical significance (p = 0.086).
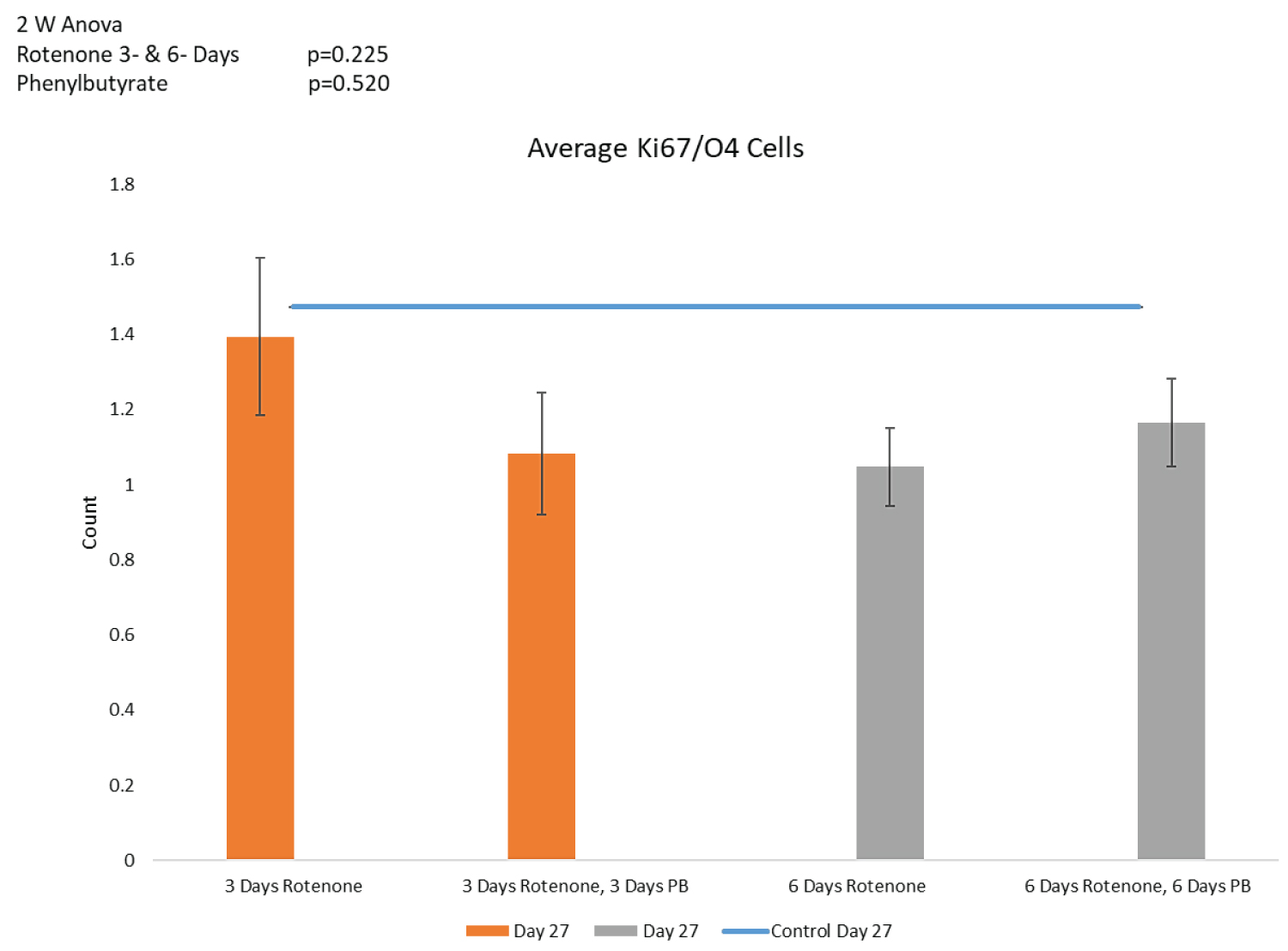
Oligodendrocyte progenitor cells (Figure 8D): In order to determine whether PB-induced regeneration of the O4+ oligodendrocytic population could be due to increased proliferation of their proliferating precursor cells, we counted the numbers of double-stained O4+/Ki67+ OPC (Figure 8D). Neither the 3- nor 6- days' supplementation with PB had a significant effect on the OPC population in the rotenone treated organoids.
Discussion
In recent years, a growing body of clinical, epidemiological and experimental studies testified to a crucial role of gestational factors in brain development and functioning in postnatal life, and affecting the vulnerability to later development of neurodegenerative disorders such as Parkinson's and Alzheimer's diseases [44,45]. Hypoxia to the fetal brain is one of the most common complications resulting in impaired circulation and brain metabolism [45]. Rotenone has been used to model hypoxia induced impairment in oxidative phosphorylation and oxidative stress. Human iPSC-derived neural stem cells subjected to daily rotenone exposures upregulated their transcription factor Nrf2 that regulates the Antioxidant-Response-Element-driven cellular defense activated by oxidative stress [46]. The Nrf2 pathway activation tends to increase with differentiation of NSCs into neurons and glia. While rotenone induced progressive increase of Nrf2 signaling there was a reduction of neurite length and mature MAP2- expressing accompanied by an induction of astrocyte reactivity neurons, all of which hinders the formation of neuronal networks [46]. In a separate study involving human iPSC, Rotenone treatment reduced the length of forming neurites as well as decreased cellular glutathione levels [47]. The effect of rotenone on oligodendrocytes populations have not been described.
In cultures of rat brain cells, oligodendrocytes were more susceptible to NO mediated damage compared to astrocytes. Amongst the brain cells oligodendrocytes have the highest metabolic rates needed to produce and maintain the myelin membranes which can represent up to 100 times the weight of the cell [48,49]. Cell respiration of oligodendrocytes is twice as high compared to neurons [50]. Oligodendrocytes have low levels of the anti-oxidant glutathione [51] and are exposed to excitotoxicity since their glutamate receptors are highly permeable to calcium [52]. With the energy breakdown, the reversal of the excitatory amino acid transporters leads to glutamate release, and glutamate-induced cytotoxicity via calcium-triggered nitrous oxide (NO) and O2 synthesis, peroxynitrite production, leading to the oligodendrocytes apoptosis [52]. White matter damage has also been associated with excitotoxicity-mediated death [53] and reactive oxygen species (ROS). OPCs are particularly sensitive to ROS as they have low antioxidant defenses and their maturation is inhibited by ROS [54,55]. Oligodendrocyte's sensitivity to ROS may be further exacerbated by their high need for iron, required for synthesis of the myelinating cholesterol [56] and high expression of the iron export transporter Ferroportin 1 [57].
In our initial tests, a high concentration of rotenone-10 μM, cerebral organoids completely deteriorated and at 2 μM concentration organoid developed but were noticeably smaller, with few or no rosettes and without clear developmental zones. At 0.2 μM, rotenone produced variable results, while 0.5 μM, rotenone produced consistently quantifiable cellular changes while preserving the general organoid structure and zones. In the present study, 3- or 6- day exposure to 0.5 μM rotenone, lead to a marked loss of oligodendrocytes. This illustrates a great utility of organoids as a model to quickly assess treatment dosages, multiple drug treatment protocols that can also be run in parallel, without the need of sacrificing large numbers of animals.
Three days rotenone exposure depleted proliferating NPC. While the organoid rosettes became smaller, the distances between individual Ki67+ cells increased. This could potentially reflect an increased NPC migration, as hypoxia has been shown to increase cell migration and induce metastasizing of human cancers [58], but could also be a result of the cell loss.
The observed rotenone induced reduction in NPC proliferation concurs with the finding that prenatal hypoxia affects the cells in brain neurogenic zones and, in particular, the levels of expression of the protein paired box 6 (Pax6). Pax6 plays an important role in neurogenesis, cell proliferation, differentiation and survival during the development of the central nervous system [45,59]. Pax6 expression is essential for controlling the balance between the proliferation and differentiation of neuronal progenitors in the cerebral cortex; intense expression of Pax6 inevitably induces neuronal differentiation in NPCs thus reducing proliferation [60].
Although, in fetuses subjected to prenatal hypoxia the levels of Pax6 were increased in the SVZ and SGZ of the hippocampal dentate gyrus but they were significantly decreased in the cerebral cortex [61]. This finding also correlates with the reduced number of neuronal cells produced in rat cortex [62].
In our experiments, when rotenone was removed from the media and the organoid allowed a 10- day post-rotenone recovery, while the number of Ki67+ cells remained reduced we observed a statistically significant increase between days 17 and 27, reaching at day 27 number similar to found in the control organoids at day 17. This partial recovery of the NPC was accompanied by the reduction in inter-cell distances indicating that the increased proliferation and reduced migration may be developmentally linked. In contrast, following a longer 6-days' mitochondrial inhibition, no recovery was observed within the span of our experiment, suggesting that the resulting changes in cell proliferation and migration increase the risk of brain malformations with the repeated or prolonged hypoxia, or other types of the mitochondrial dysfunctions. Similar observations, loss after 3-day rotenone exposure, were made for O4+ oligodendrocytes population and OPC. While there was a statistically significant decrease in O4+ cell population at both day 17 and 27 following rotenone, there was a statistically significant increase during the 10-day recovery in normal media, demonstrating the early developmental importance of oxidative phosphorylation for the development and differentiation of oligodendrocytes. In conclusion, the outcome demonstrated the importance of the mitochondrial function, oxidative phosphorylation in the developing brain NPC proliferation and migration, and oligodendrocytes development, and that the insults which impair the mitochondrial metabolism disrupt these developmental processes.
An important question concerns cellular mechanism behind oligodendrocyte recovery. Given that the NPC did not regenerate, spontaneously or after PB, the regeneration of oligodendrocytes population could be due to increased proliferation of OPC, a trend observed in the present study, or increased differentiation of the OPC to O4 expressing, nonproliferation oligodendrocytes. Consistent with the second mechanism in rat model, the hypoxia-ischemia induced a significant decrease in the number of OPCs, while the numbers of mature oligodendrocytes were affected less suggesting more efficient OPC maturation [63].
Oligodendrocyte loss due to hypoxic conditions may impair myelination as it has been observed in many congenital and acquired diseases. One such disease Periventricular leukomalacia (PVL), is a common lesion of the periventricular cerebral white matter that occurs during development, results from a hypoxic-ischemic injury and is thought to be related to the disturbance of myelin and oligodendrocyte cell lineage [64]. It was found that the declining incidence of PVL coincides with the onset of myelination in the periventricular white matter suggesting that the risk for PVL is related to the presence of late oligodendrocytes progenitors which they defined as O4+ cells [64]. In this context, the regeneration of the oligodendrocytic population following the transient mitochondrial blockade might be beneficial in combating the inflammation and increasing the chance for proper CNS myelination. Our findings may also be relevant to preterm birth associated with disrupted oxygen supply which can affect the cerebral microenvironment, cause the oxidative stress and result in myelination failure in the developing white matter. The loss of oligodendrocytes induced by oxidative stress could potentially underwrite these pathologies.
Presence of oligodendrocytes in cerebral organoids have been previously shown [65] and in Benson, et al. [41]. The present study confirmed the locations of O4+ oligodendrocytes in the intermediate and cortical zones, and expanded upon that by quantifying the oligodendrocytes population. Locations of quantified oligodendrocytes falls in the line with previously held understanding of oligodendrocytes in the human brain and animal models. Both immature and mature oligodendrocytes are present in the intermediate and cortical zones, where they provide myelinated axons from the organoid cortical areas as shown in Benson, et al. [41]. Our investigation has demonstrated the early developmental importance of oxidative phosphorylation for the development and maintenance of the oligodendrocyte population. Three days of mitochondrial inhibition halted the oligodendrocytic development in organoids but after this oxidative crisis the organoids renewed regeneration of their oligodendrocytes. However, with the longer inhibition (6-days rotenone exposure), the depressed numbers of O4+ oligodendrocytes, showed no recovery during the post-treatment time. Whether the population of mature oligodendrocytes may be restored and normal myelination may ensue will require a further longer-term investigation.
Phenylbutyrate has been shown to improve neurological status in mice after cerebral hypoxia-ischemia by alleviating cerebral infarction and neuronal apoptosis [66,67]. In our recent study, PB was shown to have beneficial effects on oligodendrocytes in a mouse model of Pyruvate Dehydrogenase Complex (PDC) deficiency which also inhibits mitochondrial functions [68]. Similar to the PDC deficient mouse, PB given following rotenone treatment significantly increased the oligodendrocyte population, appearing to induce the OPC differentiation. Unlike with the PDC deficient mouse which had a deficiency in the pyruvate dehydrogenase complex a specific target for PB action, rotenone inhibits the electron transport chain. PB's therapeutic effect mostly likely through the increase of mitochondrial fuel (acetyl CoA) but also it could be related to its function as a histone deacetylase (HDAC) inhibitor. After permanent middle cerebral artery occlusion induced hypoxia, HDAC inhibitors sodium butyrate and trichostatin A were shown to induce neurogenesis in multiple ischemic rat brain regions [69].
Fine temporal control of chromatin remodeling through ATP‐dependent chromatin remodelers and sequential histone modifiers shapes a chromatin regulatory landscape conducive to oligodendrocyte fate specification, lineage differentiation, and maintenance of cell identity. In hypoxia or direct oxidative phosphorylation blockade, ATP production is reduced, thus affecting chromatin modifications [70]. HDAC‐mediated chromatin modification to impact OPC differentiation and myelination and both processes could be rescued by HDAC inhibitors. Further investigation is needed to identify the epigenetic effects of the mitochondrial dysfunction in human brain organoid model and their modification by PB. Together, these findings provide a strong rational for the use of PB (mitochondrial agonist and HDAC-inhibitor) to mitigate white matter developmental deficits following hypoxia and other types of the mitochondrial dysfunctions.
In summary our present study using human cerebral organoid model demonstrate detrimental effects of transiently reduced mitochondrial function on the early development of the human brain involving impaired proliferation and migration of NPC, oligodendrogenesis and oligodendrocyte population. This study shows an intrinsic ability of developing brain to compensate at least partially for the transient metabolic insult which can be further enhanced by post-insult treatment with the PB. Future studies will address whether treatment with PB restores myelination and its effects on the motor functions.
References
- Domingues HS, Portugal CC, Socodato R, et al. (2016) Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol 4: 71.
- Michalski JP, Kothary R (2015) Oligodendrocytes in a nutshell. Front Cell Neurosci 9: 340.
- Simons M, Nave KA (2015) Oligodendrocytes: Myelination and axonal support. Cold Spring Harb Perspect Biol 8: a020479.
- De Juan Romero C, Borrell V (2015) Coevolution of radial glial cells and the cerebral cortex. Glia 63: 1303-1319.
- Kelava I, Lancaster MA (2016) Stem cell models of human brain development. Cell Stem Cell 18: 736-748.
- Gotz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6: 777-788.
- Lois C, Alvarez-Buylla A (1994) Long-distance neuronal migration in the adult mammalian brain. Science 264: 1145-1148.
- Menn B, Garcia-Verdugo JM, Yaschine C, et al. (2006) Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci 26: 7907-7918.
- Pencea V, Bingaman KD, Freedman LJ, et al. (2001) Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol 172: 1-16.
- Florio M, Albert M, Taverna E, et al. (2015) Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347: 1465-1470.
- Pollen AA, Nowakowski TJ, Chen J, et al. (2015) Molecular identity of human outer radial glia during cortical development. Cell 163: 55-67.
- Naruse M, Ishizaki Y, Ikenaka K, et al. (2017) Origin of oligodendrocytes in mammalian forebrains: A revised perspective. J Physiol Sci 67: 63-70.
- Richardson WD, Kessaris N, Pringle N (2006) Oligodendrocyte wars. Nat Rev Neurosci 7: 11-18.
- Spassky N, de Castro F, Le Bras B, et al. (2002) Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J Neurosci 22: 5992-6004.
- Tekki-Kessaris N, Woodruff R, Hall AC, et al. (2001) Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development 128: 2545-2554.
- Kessaris N, Fogarty M, Iannarelli P, et al. (2006) Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 9: 173-179.
- Chapman H, Waclaw RR, Pei Z, et al. (2013) The homeobox gene Gsx2 controls the timing of oligodendroglial fate specification in mouse lateral ganglionic eminence progenitors. Development 140: 2289-2298.
- Jakovcevski I, Filipovic R, Mo Z, et al. (2009) Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat 3: 5.
- Jakovcevski I, Zecevic N (2005) Olig transcription factors are expressed in oligodendrocyte and neuronal cells in human fetal CNS. J Neurosci 25: 10064-10073.
- Rivkin MJ, Flax J, Mozell R, et al. (1995) Oligodendroglial development in human fetal cerebrum. Ann Neurol 38: 92-101.
- Jakovcevski I, Zecevic N (2005) Sequence of oligodendrocyte development in the human fetal telencephalon. Glia 49: 480-491.
- Rakic S, Zecevic N (2003) Early oligodendrocyte progenitor cells in the human fetal telencephalon. Glia 41: 117-127.
- Leong SY, Rao VT, Bin JM, et al. (2014) Heterogeneity of oligodendrocyte progenitor cells in adult human brain. Ann Clin Transl Neurol 1: 272-283.
- van Tilborg E, de Theije CGM, van Hal M, et al. (2018) Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 66: 221-238.
- Rosko L, Smith VN, Yamazaki R, et al. (2019) Oligodendrocyte bioenergetics in health and disease. Neuroscientist 25: 334-343.
- Schoenfeld R, Wong A, Silva J, et al. (2010) Oligodendroglial differentiation induces mitochondrial genes and inhibition of mitochondrial function represses oligodendroglial differentiation. Mitochondrion 10: 143-150.
- El Waly B, Macchi M, Cayre M, et al. (2014) Oligodendrogenesis in the normal and pathological central nervous system. Front Neurosci 8: 145.
- Hesse A, Wagner M, Held J, et al. (2010) In toxic demyelination oligodendroglial cell death occurs early and is FAS independent. Neurobiol Dis 37: 362-369.
- Lindner M, Fokuhl J, Linsmeier F, et al. (2009) Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci Lett 453: 120-125.
- Mason JL, Toews A, Hostettler JD, et al. (2004) Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am J Pathol 164: 1673-1682.
- Betarbet R, Sherer TB, MacKenzie G, et al. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301-1306.
- Radad K, Rausch WD, Gille G (2006) Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int 49: 379-386.
- Li N, Ragheb K, Lawler G, et al. (2003) Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 278: 8516-8525.
- Giordano S, Lee J, Darley-Usmar VM, et al. (2012) Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS One 7: e44610.
- Xiong ZK, Lang J, Xu G, et al. (2015) Excessive levels of nitric oxide in rat model of Parkinson's disease induced by rotenone. Exp Ther Med 9: 553-558.
- Gao F, Chen D, Hu Q, et al. (2013) Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS One 8: e72046.
- Lancaster MA, Renner M, Martin CA, et al. (2013) Cerebral organoids model human brain development and microcephaly. Nature 501: 373-379.
- Stachowiak EK, Benson CA, Narla ST, et al. (2017) Cerebral organoids reveal early cortical maldevelopment in schizophrenia-computational anatomy and genomics, role of FGFR1. Transl Psychiatry 7: 6.
- Chuye LB, Dimitri A, Desai A, et al. (2018) Brain organoids: Expanding our understanding of human development and disease. Results Probl Cell Differ 66: 183-206.
- Narla ST, Lee YW, Benson CA, et al. (2017) Common developmental genome deprogramming in schizophrenia - role of integrative nuclear fgfr1 signaling (INFS). Schizophr Res 185: 17-32.
- Benson CA, Powell HR, Liput M, et al. (2020) Immune factor, TNFα, disrupts human brain organoid development similar to schizophrenia-schizophrenia increases developmental vulnerability to TNFα. Front Cell Neurosci 14: 233.
- Arnold B, Cassady SJ, VanLaar VS, et al. (2011) Integrating multiple aspects of mitochondrial dynamics in neurons: Age-related differences and dynamic changes in a chronic rotenone model. Neurobiol Dis 41: 189-200.
- Shimada IS, LeComte MD, Granger JC, et al. (2012) Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J Neurosci 32: 7926-7940.
- Faa G, Marcialis MA, Ravarino A, et al. (2014) Fetal programming of the human brain: Is there a link with insurgence of neurodegenerative disorders in adulthood? Curr Med Chem 21: 3854-3876.
- Nalivaeva NN, Turner AJ, Zhuravin IA (2018) Role of prenatal hypoxia in brain development, cognitive functions, and neurodegeneration. Front Neurosci 12: 825.
- Pistollato F, Canovas-Jorda D, Zagoura D, et al. (2017) Nrf2 pathway activation upon rotenone treatment in human iPSC-derived neural stem cells undergoing differentiation towards neurons and astrocytes. Neurochem Int 108: 457-471.
- Neely MD, Davison CA, Aschner M, et al. (2017) From the cover: Manganese and rotenone-induced oxidative stress signatures differ in iPSC-derived human dopamine neurons. Toxicol Sci 159: 366-379.
- Connor JR, Menzies SL (1996) Relationship of iron to oligodendrocytes and myelination. Glia 17: 83-93.
- Morell P, Toews AD (1984) In vivo metabolism of oligodendroglial lipids, Springer, Boston, MA.
- Cammer W (1984) Carbonic anhydrase in oligodendrocytes and myelin in the central nervous system. Ann N Y Acad Sci 429: 494-497.
- Thorburne SK, Juurlink BH (1996) Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem 67: 1014-1022.
- Benarroch EE (2009) Oligodendrocytes: Susceptibility to injury and involvement in neurologic disease. Neurology 72: 1779-1785.
- Matute C, Alberdi E, Domercq M, et al. (2007) Excitotoxic damage to white matter. J Anat 210: 693-702.
- French HM, Reid M, Mamontov P, et al. (2009) Oxidative stress disrupts oligodendrocyte maturation. J Neurosci Res 87: 3076-3087.
- Volpe JJ, Kinney HC, Jensen FE, et al. (2011) The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int J Dev Neurosci 29: 423-440.
- Badaracco ME, Siri MV, Pasquini JM (2010) Oligodendrogenesis: The role of iron. Biofactors 36: 98-102.
- Burdo JR, Menzies SL, Simpson IA, et al. (2001) Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J Neurosci Res 66: 1198-1207.
- Doronkin S, Djagaeva I, Nagle ME, et al. (2010) Dose-dependent modulation of HIF-1alpha/sima controls the rate of cell migration and invasion in Drosophila ovary border cells. Oncogene 29: 1123-1134.
- Simpson TI, Price DJ (2002) Pax6; a pleiotropic player in development. Bioessays 24: 1041-1051.
- Osumi N, Kikkawa T (2013) The role of the transcription factor Pax6 in brain development and evolution: Evidence and Hypothesis. In: T Yamamori, R Kageyama, Cortical development. (edn), Springer, Tokyo, Japan, 43-61.
- So K, Chung Y, Yu SK, et al. (2017) Regional immunoreactivity of Pax6 in the neurogenic zone after chronic prenatal hypoxia. In Vivo 31: 1125-1129.
- Vasilaki DS, Tumanova NL, Zhuravin IA (2008) Structural changes in the neocortex nervous tissue in rat ontogenesis after hypoxia at various terms of embryogenesis. Journal of Evolutionary Biochemistry and Physiology 44: 304-315.
- Ziemka-Nalecz M, Janowska J, Strojek L, et al. (2018) Impact of neonatal hypoxia-ischaemia on oligodendrocyte survival, maturation and myelinating potential. J Cell Mol Med 22: 207-222.
- Back SA, Luo NL, Borenstein NS, et al. (2001) Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 21: 1302-1312.
- Renner M, Lancaster MA, Bian S, et al. (2017) Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J 36: 1316-1329.
- Iannitti T, Palmieri B (2011) Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 11: 227-249.
- Qi X, Hosoi T, Okuma Y, et al. (2004) Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol 66: 899-908.
- Freedman D, Klejbor I, Mahmood S, et al. (2020) Loss of Oligodendrocytes in Mouse Model of Pyruvate Dehydrogenase Complex Deficiency and Partial Reversal by Phenylbutyrate Treatment. Transl Neurosci Res Rev 3: 53-61.
- Kim HJ, Chuang DM (2014) HDAC inhibitors mitigate ischemia-induced oligodendrocyte damage: Potential roles of oligodendrogenesis, VEGF, and anti-inflammation. Am J Transl Res 6: 206-223.
- Gregath A, Lu QR (2018) Epigenetic modifications-insight into oligodendrocyte lineage progression, regeneration, and disease. FEBS Lett 592: 1063-1078.
Corresponding Author
Ewa K Stachowiak, PhD, Department of Pathology and Anatomical Sciences, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY, 14203 USA; Western NY Stem Cell Center, Stereology Laboratory, State University of New York at Buffalo, Buffalo, NY, 14203 USA
Michal K Stachowiak, PhD, Department of Pathology and Anatomical Sciences, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY, 14203 USA; Western NY Stem Cell Center, Stereology Laboratory, State University of New York at Buffalo, Buffalo, NY, 14203 USA
Copyright
© 2020 Freedman D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.