Loss of Oligodendrocytes in Mouse Model of Pyruvate Dehydrogenase Complex Deficiency and Partial Reversal by Phenylbutyrate Treatment
Abstract
Human pyruvate dehydrogenase complex (PDC) deficiency caused by mutations of the pyruvate dehydrogenase component (PDH) of the pyruvate dehydrogenase complex (PDC) is associated with the loss of myelin in the cortex and subcortical brain regions. Similar changes occur in the heterozygous PDH+/- mouse model of a partial PDC deficiency. In order to investigate the underlying mechanisms, quantitative stereological analysis of O4 expressing oligodendrocytes (OLG), double stained Ki67+/O4+ immature proliferating OLG Progenitor Cells (OPCs) and overall numbers of the proliferating cells (Ki67+) were analyzed in the brains of 35-day-old PDH+/- and control PDH+/+ mice. ANOVA showed an overall depletion of all cell populations in analyzed brain regions in PDH+/- mice. As a potential treatment for PDC insufficiency we tested phenylbutyrate (PB), an inhibitor of PDH kinases, to increase the residual PDC activity. In PDH+/+ mice, daily (from postnatal day 2-35) single injections of PB produced no effect, though in the PDH+/- mice, PB had an overall increasing effect on O4 OLG with significant increases recorded for SVZ and SGZ. PB increased and restored depleted OPCs in PDH+/- mice to the levels found in PDH+/+. In summary, loss of O4 OLG in developing PDC-deficient mice and their PB-induced recovery appear to reflect changes in their proliferating progenitors and may be beneficial for the treatment of PDC deficiency in children affected with mutations in the α subunit of PDH.
Keywords
Pyruvate Dehydrogenase complex deficiency, Oligodendrogenesis, Phenylbutyrate
Abbreviations
CA: Cornu ammonis; DG: Dentate gyrus; NO: Nitric Oxide; OLG: Oligodendrocytes; OPCs: Oligodendrocyte Progenitor Cells; PB: Phenylbutyrate; PC: Purkinje Cells; PDC: Pyruvate Dehydrogenase Complex; PDH: Pyruvate Dehydrogenase; PDK: Pyruvate Dehydrogenase Kinase; PDP: Pyruvate Dehydrogenase Phosphatase; ROI: Region of Interest; ROS: Reactive oxygen species; SVZ: Subventricular zone
Introduction
Pyruvate Dehydrogenase Complex (PDC), a mitochondrial matrix enzyme, converts pyruvate, the end product of cytoplasmic glycolysis, into acetyl-coenzyme A which is further metabolized in the citric acid cycle. PDC consists of a thiamine diphosphate-dependent pyruvate dehydrogenase (PDH), a heterotetramer of 2α and 2β subunits (α2β2); dihydrolipoamide acetyltransferase, and FAD-containing dihydrolipoamide dehydrogenase (E3), which is integrated into the complex by an E3-binding protein (E3BP) [1-4].
PDC is regulated through phosphorylation of the α-subunit of PDH by four pyruvate dehydrogenase kinase (PDK) isoforms at three specific serine residues (Site 1: Ser264; Site 2: Ser271; and Site 3: Ser203) [5]. Phosphorylation at any one of these sites renders the complex inactive. PDK isoforms exhibit different specificities for the three PDHα serine sites. Sites 1 and 2 are phosphorylated by all four isoforms whereas site 3 is only modified by PDK1 [6-8]. PDK isoforms have different tissue expression; PDK1 is highly expressed in heart, PDK2 is ubiquitously expressed, PDK3 has a relatively limited tissue distribution (mostly in testis and to a lesser extent in brain, lung, and kidney), and PDK4 is expressed in heart and skeletal muscle [8,9]. PDK2 is expressed at higher levels compared to other iso-enzymes, suggesting that it may be the major isoform responsible for regulation of PDC enzyme activity [8,9]. Dephosphorylation of the PDHα by pyruvate dehydrogenase phosphatases 1 and 2 restores PDC activity [8,10,11].
PDC deficiency is one of the most common inborn errors in the mitochondrial energy metabolism. It may be due to mutations in PDHA1, PDHB, PDHX, DLAT, and DLD genes encoding PDH-α, PDH-β, dihydrolipoamide dehydrogenase-binding protein, dihydrolipoamide acetyltransferase, and dihydrolipoamide dehydrogenase proteins, respectively or due to mutations of the pyruvate dehydrogenase phosphatases 1 (PDP1) gene encoding pyruvate dehydrogenase phosphatase 1 [10,11]. In one of the largest studies performed on PDC deficiency which reviewed data, from 371 patients, Patel, et al. found that 84% of mutations are located on the PDHA1 gene resulting in X-linked heterogeneous condition. Missense mutations were more prevalent in males while females presented more frameshift mutations [11]. Of the missense mutations, 77% were localized on exons 1-9 while 86.5% of frameshift mutations were localized on exons 10 and 11 [11].
Clinically the disease presents itself in a myriad of ways largely affecting the brain: Developmental delay, mental or psychomotor retardation, microcephaly, ventriculomegaly and hypogenesis or agenesis of the corpus callosum [11]. Other reports also found dilation of the lateral cerebral ventricles and underdevelopment of large white matter structures such as the corpus callosum, the pons and pyramids. Atrophy or neuronal loss combined with gliosis in the cortex and basal ganglia, thalamus, hypothalamus, and cerebellum were also described [12,13]. Since most mutations occur on the X linked PDHA1 gene, males are less likely to survive infancy.
There are currently three strategies used to treat PDC-deficient patients (i) A ketogenic diet, (ii) Supplementation of high doses of thiamin [14,15], and (iii) Treatment with dichloroacetate, which has been shown to increase PDC activity by inhibiting the PDKs but has been associated with hepatocellular and peripheral nerve toxicity [8,16]. All three therapies have been associated with reduction of blood lactate and some improvement clinical manifestations but prevention of disease development or its reversal has yet to be accomplished.
A new potential treatment could involve a stimulation of acetyl-CoA production from glucose by increasing activity of the residual PDH and hence PDC. Since PDH activity is inhibited by its phosphorylation, the use of PB, an inhibitor of PDK activity, which increases the conversion of an 'inactive' PDC to 'active' PDC, could restore production of the oxidative energy from the pyruvate [8,10,17,18]. PB has recently been shown to inhibit PDH kinases, resulting in reduction in the phosphorylated form of PDHα protein (thus increasing PDC activity) in tissues from normal adult mice injected with this inhibitor [17], in fibroblasts from PDC-deficient patients [17] and also corrected the morphological, locomotor, and biochemical abnormalities in the noam631 zebrafish model of PDC deficiency [19,20]. PB administration reduced or prevented systemic lactic acidosis induced by partial hepatectomy, heart ischemia, septic shock, and stroke [21,22]. A protective role of PB was shown during cerebral ischemia [23,24].
While PB is converted/oxidized in vivo into phenylacetate by β-oxidation, it has been shown that PB and not its bi-product phenylacetate is effective in increasing PDC activity [8,18,25]. Using an in vivo adult mouse model, western blot analysis of freshly isolated brain mitochondria documented a statistically significant 3-fold decrease in the phosphorylated PDHα and an increase in PDC enzymatic activity in the mouse brain following 3 days of 250 mg/kg/day PB treatment, measured 1.5 hours after the last administration [17].
Using murine model of systemic PDC deficiency with interrupted X-linked Pdha1 gene (encoding the α subunit of PDH), Pliss, et al. [26] showed alterations in several structures in white and gray matters in 35-day-old PDC-deficient females. Reduction in total cell number and reduced dendritic arbors in Purkinje neurons were observed in PDC-deficient females. Furthermore, cell proliferation, migration and differentiation into neurons were also observed [26].
In a recent study, Klejbor, et al. [27] also used female mice with a conditional Cre-loxP mutation [28] introduced into the Pdha1 gene located on chromosome X [29]. The mice were generated by breeding the transgenic males {B6.Cg[SJL]-TgN[NesCre]1Kln}(Crebr) harboring a Cre transgene driven by the nestin promoter with the Pdha1-floxed females from B6 genetic background. Female control Cre-positive_(PDH+/+) and PDH-deficient progenies (PDH+/-) were injected intraperitoneally with PB (250 mg/kg body weight), or saline [17] once a day starting from postnatal day 2 to 35. Histological analysis of cerebellum verified the reduction in the PC density and showed increased cross-sectional area of the individual PC in 35-day-old mice. Daily administration of PB reduced changes in the density and soma size in PC populations in the areas of cerebellum in which the PC neurogenesis continues postnatal [27]. In the present study, we used these PB- or saline-treated PDH+/+ or PDH+/- mice to identify the mechanisms underlying the agenesis of the brain white matter, observed in female PDC-deficient patient and in PDC- deficient P35 female mice [26,30,31]. To identify the mechanisms underlying of white matter agenesis, our current investigation has focused on brain myelinating cells, oligodendrocytes and their proliferating precursors (Oligodendrocyte Precursor cells, OPCs).
Amongst the brain cells, OLGs show the highest metabolic rates needed to produce and maintain the high volume of myelin membranes which can represent up to 100 times the weight of the cell [32,33]. Cell respiration in OLGs has been reported to be twice as high compared to neurons [34]. OLGs have also been shown to have low levels of glutathione, a major anti-oxidant [35].
OLGs are also exposed to excitotoxicity since their glutamate receptors are highly permeable to calcium [36]. With the energy breakdown, the reversal of the excitatory amino acid transporters leads to glutamate release, and glutamate-induced cytotoxicity via calcium-triggered nitric oxide synthesis, peroxynitrite (an oxidant and nitrating agent) production, leading to the OLGs apoptosis [36]. In rat brain cultures, OLGs were more susceptible to nitric oxide-mediated damage compared to their glial counterparts. In addition, activated microglial cells appeared to produce sufficient nitric oxide to lyse the co-cultured OLGs and this could be prevented with antagonists of nitric oxide production [37].
White matter damage has also been associated with PDC deficiency [38] as well as excitotoxicity-mediated death [39] and reactive oxygen species (ROS). OPCs have been shown to be particularly sensitive to ROS as they have low antioxidant defenses and their maturation is inhibited by ROS [40,41]. OLGs sensitivity to ROS may be further exacerbated by their high need for iron, required for synthesis of the myelinating cholesterol [42]. Strong immunostaining for iron export transporter Ferroportin 1, suggesting an active protective iron efflux, has been shown in subcortical OLGs [43].
In the present study we show that the population of OLGs is severely affected by PDH+/- mutation but may be partially restored by PB treatment which reconstitutes their proliferating OPCs.
Materials and Methods
Animal strains used
The mice used in the present study were the female progeny with heterozygous PDH+/- mutation and control PDH+/+ mice weaned onto standard rodent laboratory diet and water on postnatal day 21. The mice were generated and bred as reported in [27]. Mice genotypes were verified by PCR of tail DNA using specific primer sets and conditions as previously described and additionally verified by PCR of the brain DNA [27]. All animal procedures were approved by the University at Buffalo Institutional Animal Care and Use Committee, protocol # BCH11064Y.
The four groups studied were generated in our recent study [27]: (i) Control PDH+/+ saline- injected, (ii) PDH+/+ PB-injected, (iii) PDH+/- saline-injected, and (iv) PDH+/- PB-injected. Mice were injected with sterile saline or PB dissolved in sterile saline (250 mg/kg body weight). Briefly, mice received daily injections starting from postnatal day 2 until day 35. Approximately 18 h after the last injection mice were anesthetized, euthanized by transcardial perfusion and the brains were fixed as described in [27]. After the cerebella were removed for the study described in [27], telencephalic regions were dissected including cortical region, subventricular zone and hippocampus for the present investigation.
The average body weight of the saline-injected (8.86 +/- 1.52 g, n = 5) and PB-injected (10.42 +/- 1.03 g, n = 4) PDH+/- mice were not significantly different from each other but were significantly lower than the weighs of saline- (14.1 ± 0.3 g) and PB-injected (14.1 ± 0.3 g) PDH+/+ mice reported in [27].
Tissue preparation
Cryostat (Microm HM 505 N) coronal sections were cut at 40 μm thickness. The start and end points for each region of interest (ROI) in the study, was determined using Paxinos' Mouse Atlas: For SVZ (bregma +1.18 mm - bregma -0.22 mm), hippocampal SGZ (bregma -0.94 mm - bregma -4.16 mm) and Cortex (bregma +1.18 mm - bregma -0.22 mm). All sections were serially transferred into 48-well plates containing a mixture of anti-freezing solution with 30% ethylene glycol, 25% glycerol and 45% 1× phosphate buffered saline (PBS). The prepared material yielded approximately 30 sections per ROI (SVZ, SGZ, and Cortex). The first section was selected at bregma 1.10 mm for both SVZ and Cortex while bregma -1.06 mm was used for hippocampal SGZ. Thereafter every sixth section was selected for immunostaining for a total of five sections per ROI (cortex, SVZ, SGZ).
Immunostaining and stereological analyses
Sections were rehydrated in PBS, twice for 10 minutes, and post-fixed with 4% Paraformaldehyde for 10 minutes. Following fixation, sections were washed twice in PBS (20 minutes) and then permeabilized with 0.5% Triton-X-100 (15 minutes). After two washes with PBS (20 minutes), sections were blocked with protein block (Biogenex) (1 hour at room temp) and incubated with primary antibody diluted in Antibody Dilutent (IHC World) overnight in 4 °C. After 3 washes in 1 × PBS sections were next incubated with secondary antibody diluted in Antibody Diluent (IHC World) for 2 hours. For double staining, the second primary antibody was added after completing 1st stain and the procedure followed the same protocol as mentioned above. After completion of immunostaining slides were cover-slipped with Fluoromount II with DAPI (Electron Microscopy Science).
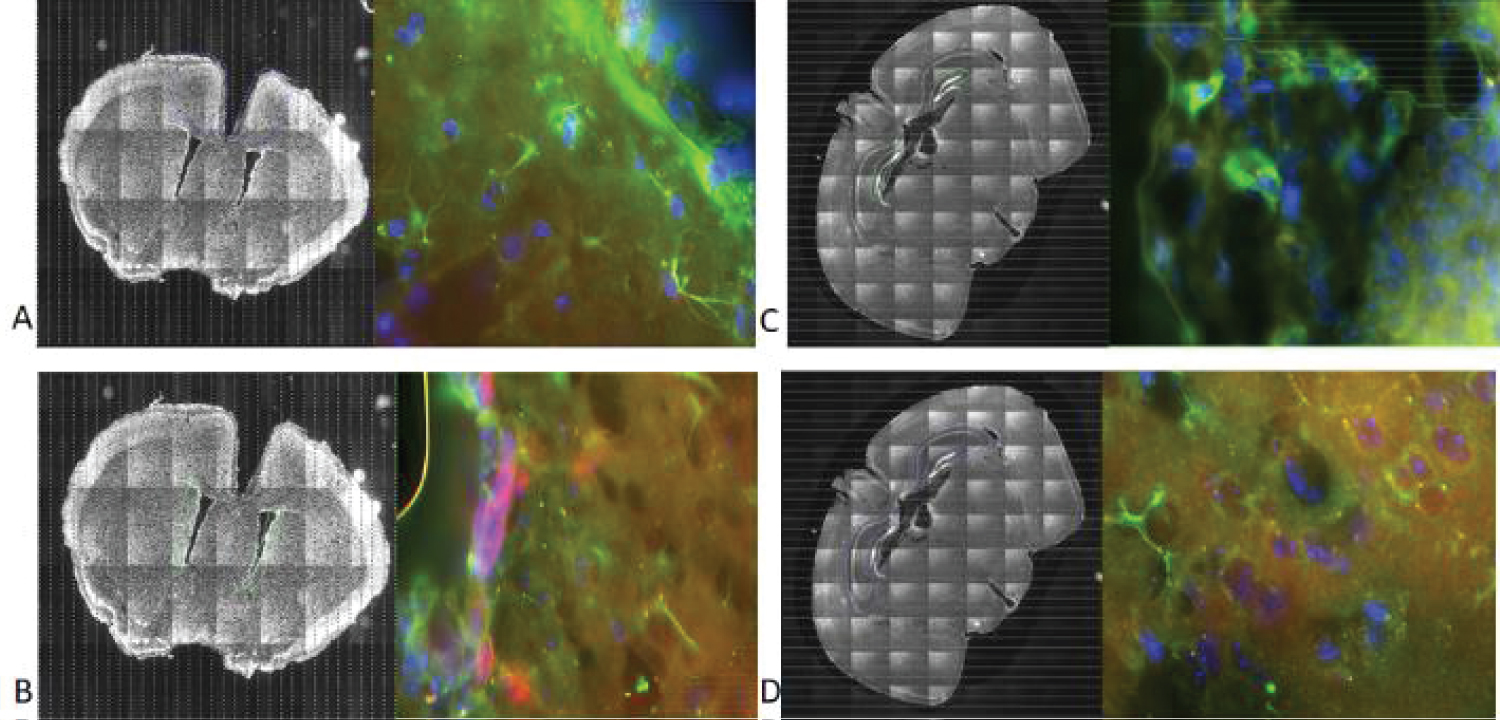
Mitotically active cells were detected by immunostaining using rabbit polyclonal (Cat #: ab15580, abeam) anti-Ki67 antibody and the polyclonal secondary antibody at a dilution of 1:1000 (Cat. # A11011, Life Technologies- Molecular Probes). Staining for Oligodendrocyte Marker O4 expressed by oligodendrocytes, be but also present in dividing OPCs was performed using Monoclonal Antibody IgM at dilution 1:330 (Cat #: MO15002.9, Neuromics) followed by Alexa Fluor® Goat pAb anti-Mouse IgM (Cat # ab150121, abcam) secondary antibody. The single or double stained sections were mounted on microscopic slides using Fluorogel II with DAPI (Cat. # 17985-50, Electron Microscopy Sciences) for visualization of nuclei.
Imaging was performed on an Olympus BX61 fully automated fluorescence microscope equipped with UIS2 optical system and with 25 mm motorized focus/vertical stage movement.
Images were analyzed using NewCast software (Visiopharm, Denmark). Regions of interests (ROI) were outlined or masked using Paxinos' mouse atlas as a reference. A subset of the ROI was then sampled using Optical Fractionator. In Optical Fractionator, randomly selected and equidistant field of visions (FOV) were sampled. The sampled areas were determined based on cell density in order to avoid overlapping and they were as followed: 55% of SVZ, 45% of SGZ (DG and CA) and 25% of the cortex. The FOVs were viewed at a set area-sampling fraction of the masks using NewCast sampling software at 60× magnification for all areas. FOVs consisted of stacks of images taken through the z-axis of the ROI at a set interval (step size) of 2 micrometers for all areas. Care was taken to ensure that the entire depth of the section was imaged. The emission wavelength was set in the following order while digitizing the tissues: 603 nm for Ki67, 530 nm for O4, and 461 nm for DAPI.
From each mouse brain structure, four sections were analyzed generating four ROIs. The numbers of individual ROIs were averaged for each mouse brain structure (n = 1). For each of the three experimental conditions (namely saline-injected PDH+/+ control mice, PB-injected PDH+/+ control mice, saline-injected mutant PDH+/- mice), 5 mice were analyzed (n = 5). In the PB-injected PDH+/- mutant mice group, 4 mice were analyzed (n = 4). Statistical analysis, both one- way and two-way ANOVA, were performed using IBM SPSS (University at Buffalo) software. Differences were considered significant with p ≤ 0.05.
Results
Effect of PDH+/- mutation and PB on cell proliferation
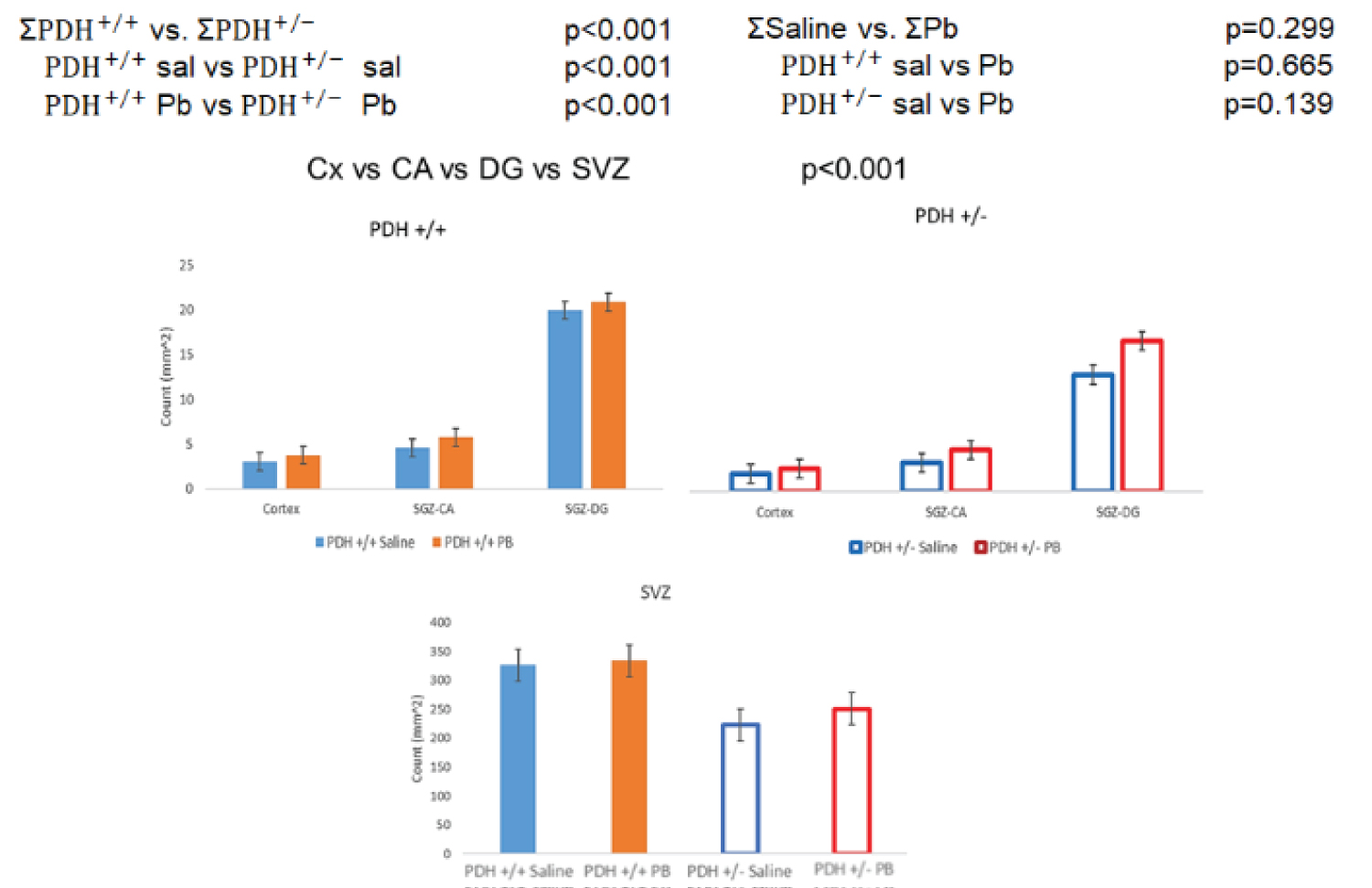
To determine the effects of PDC insufficiency on cell proliferation in brain cortex and in neurogenic SVZ and SGZ, in which proliferating population is represented by NPC, we analyzed expression of Ki67 immunopositive cells. Ki67 is expressed at different phases of the mitotic cycle and its expression is turned off at the exit from the cycle into the G1 phase [44]. Four groups of mice were analyzed: i) Saline-injected PDH+/+ control-mice (ii) PB-injected PDH+/+ mice, (iii) Saline-injected mutant PDH+/- mice and (iv) PB-injected PDH+/- mice (for details see [27] the Materials and Methods). Examples of Ki67 staining and O4 stains are shown in Figure 1.
PDH+/- mutation depleted population of proliferating cells
Results of stereological counting of Ki7+ cells were analyzed using two-way-ANOVA and are shown in Figure 2. The combined analysis of all brain regions, SVZ, SGZ, and Cortex demonstrated an overall reducing effect of PDH+/- mutation on the population of proliferating (Ki67+ cells) independent of the type of, saline or PB injections. No significant effect of the PB injection compared to the saline injection was found.
Effect of PDH+/- mutation and PB on O4 OLG population
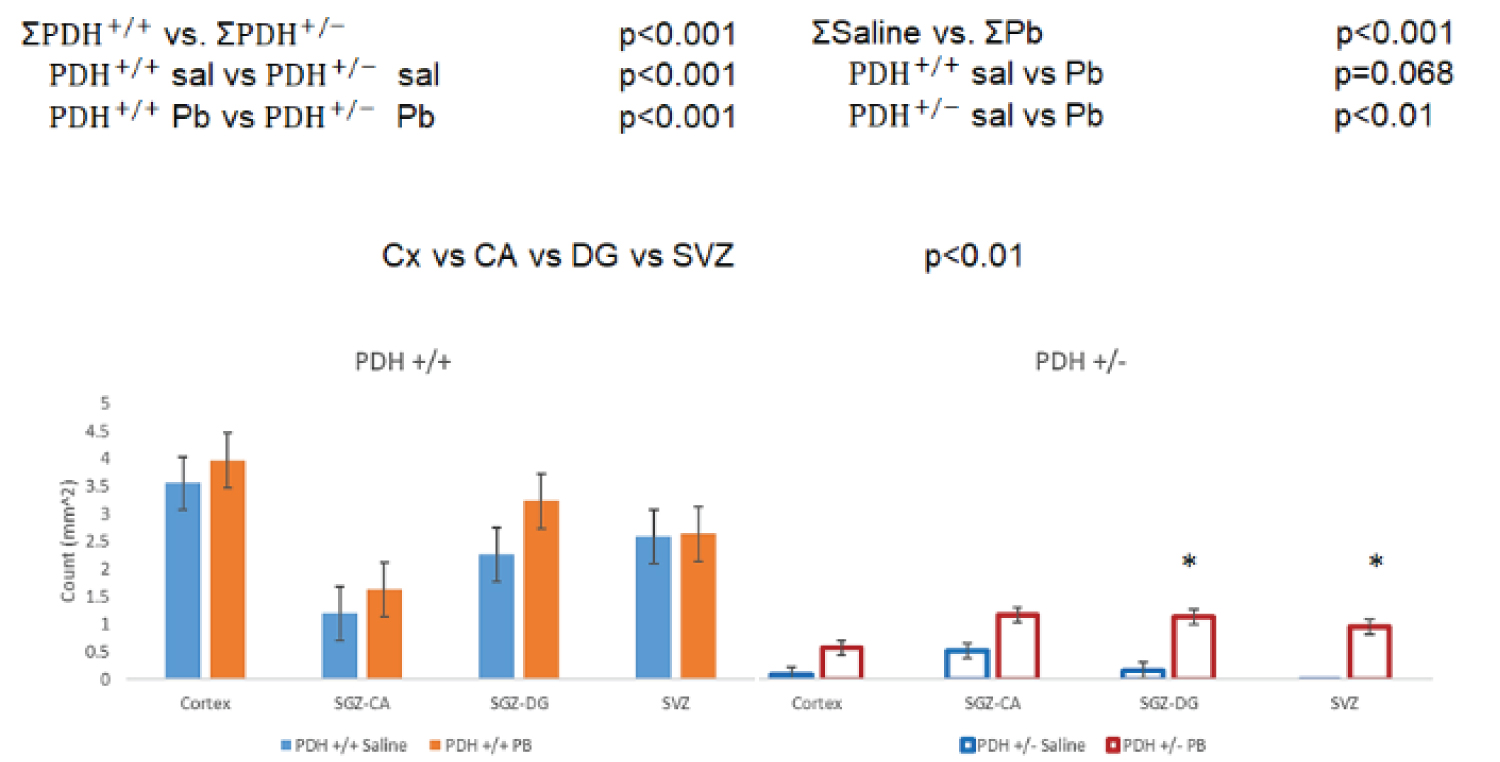
The O4 antibody reacts with sulfated galactosylcerebroside, sulfatide. And thereby stains late oligodendrocyte progenitors as well as oligodendrocytes that have entered terminal differentiation [45]. The O4 immunostaining revealed presence of OLG cells in all brain areas examined with greatest densities observed in the neocortex and the lowest in the hippocampal CA. Two-way ANOVA of all brain regions showed an overall reducing effect of PDH+/- mutation on the population of O4+ OLG which was significant for both saline and PB-treated mice. Also, treatment with PB had an overall significant effect on the O4 population. In the PDH+/+ mice, daily injections of PB produced no effect on O4 or OPCs. In contrast, in the PDH+/- mice, PB had an overall increasing effect on O4 OLG with significant increases recorded for SVZ. In PDH+/+ mice, daily injections of PB produced no effect on O4 or OPCs. In contrast, in the PDH+/- mice, PB had an overall increasing effect on O4 OLG with significant increases recorded for SVZ and SGZ-DG (Figure 3).
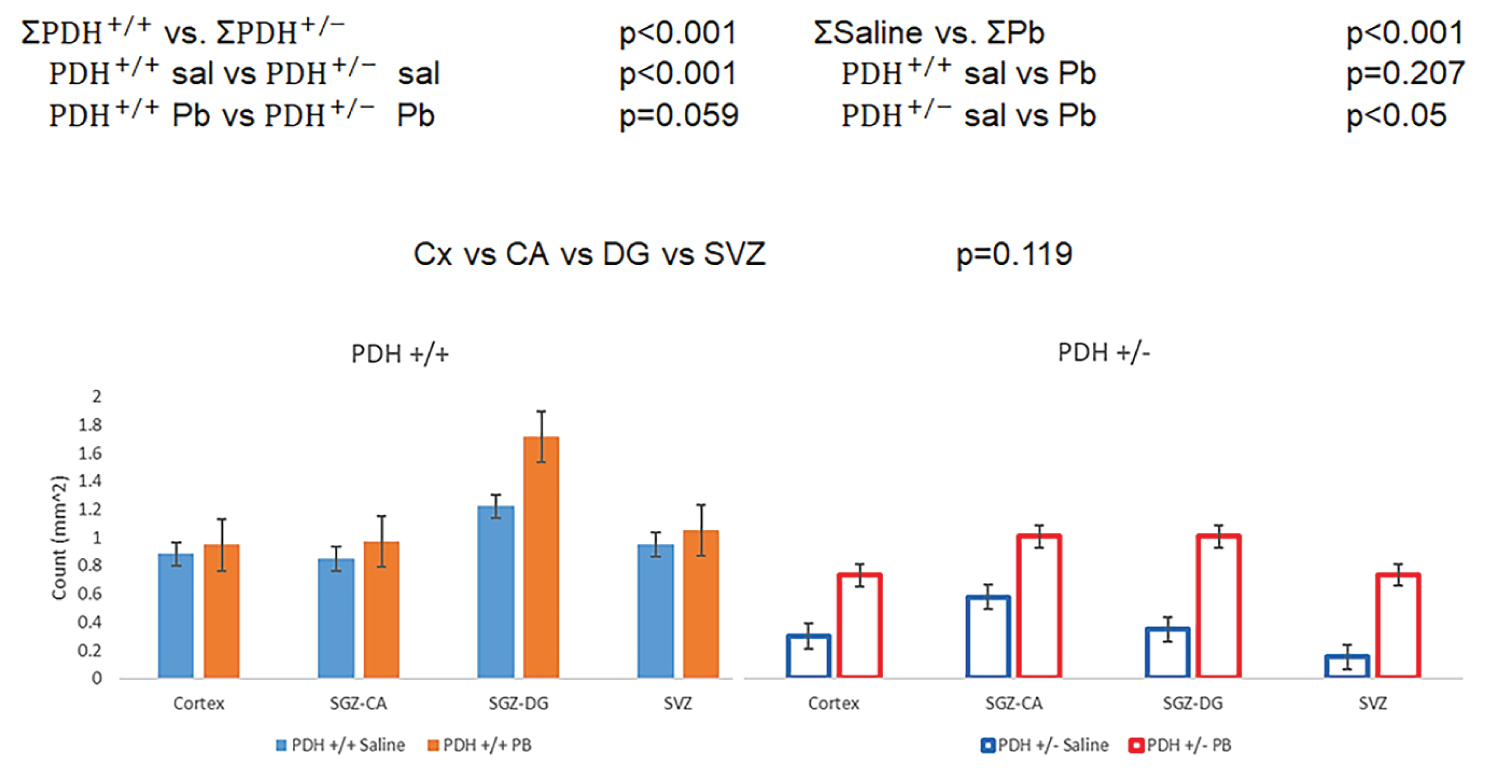
PB partially restored the numbers of O4+ OLG in PDH+/- mice in which the mutation depleted the O4 cells. While in control mice the O4 OLG population appeared increased after the PB treatment, this change did not attain the statistically significant level (Figure 3). The analysis of the relatively small population of double labeled Ki67+/O4+ provided insight into immature, still proliferating, population of oligodendrocytes referred to as OPCs (Figure 4).
PDH+/- mutation produced an overall loss of OPCs which was statistically significant for both the saline and the PB-treated mice. While PB had an overall significant effect, the drug induced increase was reached the significant levels only in the mutant PDH+/- mice in which it restored the OPCs population to the levels found in PDH+/+ mice.
Discussion
In 35-day-old mice the PDH+/- mutation reduced the overall populations of OLG, their precursor OPCs as well as total cell proliferation in the analyzed brain regions. The population of OLG in PDC deficient, PDH+/- mice was upregulated and the OPCs were restored to the control levels by PB. These findings are consistent with the PB ability to inhibit all PDK isoforms 1-3; with the strongest inhibition of brain isoforms PDK 2 and 3, likely the main target for PB in vivo [8].
Within the neurogenic niches SVZ and SGZ-DG the proliferating (Ki67+ cells) largely represents multipotent population of NPC [46,47]. In the present study, these cells were found to be depleted by PDH+/- mutation but were not restored or in any way significantly affected by PB. In contrast, the population of proliferating OLG lineage-committed OPCs (Ki67+/O4+) which constituted a small fraction of the NPC (0.3% in SVZ and 5% in SGZ DG) was upregulated and reconstituted by PB. In the non-neurogenic area, the neocortex, and in hippocampal archicortex, there were fewer proliferating cells and OPCS constituted 27% (cortex) and 16% (CA) of all mitotic cells. Similarly, as in the neurogenic niches, however, only the OPCs were upregulated by PB indicating that the drug was effective specifically for the restitution of the OLG lineage progenitor cells.
The mechanism by which PB produces its restorative effects were illustrated in the studies of Ferriero, et al. which demonstrated PB-induced marked dephosphorylation of the PDHα in the brain, liver and the muscle tissues [17] and further discussed in [48]. PB is known also as a histone deacetylase inhibitor and this could potentially play a role in upregulation of OLG and OLG progenitors in the current study of PDC-deficient mice. Histone deacetylase inhibitors have been shown to induce differentiation in a variety of cell types [18]. Additionally, in disease pathologies such as multiple sclerosis, PB has been shown to reduce neuroinflammation [18,49] and overall disease process while in Huntington's disease, PB has been shown to significantly reduce the effect of gross brain atrophy and ventricular enlargement as well as neuronal atrophy [18,50]. In addition, lactic acidosis, a major general manifestation of mitochondrial disease, has been shown to increase in the brain in PDC [51]. This excessive amount the lactic acid may lead to structural alterations and impaired cell survival.
In mice, PB has been shown to prevent systemic lactic acidosis induced by partial hepatectomy which may show another mechanism of the therapeutic benefit of PB. In a recent study the Funfschilling, et al. [47] looked at the nuclear COX10 gene which is essential for the electron transport chain in mitochondria. Mice with a mutant COX10 gene, the mature oligodendrocytes were still able to maintain myelin and survived by shifting to aerobic glycolysis. However, in vitro cultured oligodendrocytes, which correspond to pre-myelinating oligodendrocytes were sensitive to the COX inhibitor azide. Thus, it appears that the metabolic properties of oligodendrocytes lineage cells change during maturation [52].
In adult mice, the PDH knock-down in oligodendrocytes or in Schwann cells did not affect the existing myelination of their targets: Spinal cord neurons or the peripheral sciatic nerve, respectively [53]. Thus, the loss of OLG caused by PDH+/- mutation observed in the present developmental study, appears to be associated specifically with the CNS development. Its primary cause may be the loss of OPCs as both the PDH+/- induced losses and the PB-induced recovery of the O4 OLG were accompanied by parallel changes in the OPCs. Our finding prompt further inquiry into potential PB-mediated regeneration of the myelin wrappings in the brain neurons during development.
Author Contributions
DF performed brain sectioning, immunohistochemistry, microscopy, stereological and statistical analyses and wrote the manuscript. EKS and MKS initiated current oligodendrocyte study, designed immunohistological studies and supervised analyses, co-wrote and edited manuscript. MSP designed animal breeding strategy, genotyping, and phenylbutyrate treatment, and supervised analyses described in [27], and discussed and contributed to editing the current study manuscript. SM and IK bred PDH+/- and PDH+/+ mice analyzed DNA, performed mice injections, collected body weights, perfused mice, dissected brains and provided fixed brains from the study described in [27] and contributed to the editing of the current manuscript.
Acknowledgements
Study was supported by National Science Foundation (CBET-1555720; CBET-1706050), and by Patrick P. Lee Foundation provided to MKS. Generation and treatment of mice [27] was supported by US Public Health Service Grant R21NS093264 to MSP, MKS and EKS.
Competing Interest
The authors declare no competing interests.
References
- Reed LJ (2001) A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem 276: 38329-38336.
- Patel MS, Roche TE (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J 4: 3224-3233.
- Harris RA, Bowker-Kinley MM, Huang B, et al. (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 42: 249-259.
- Roche TE, Baker JC, Yan X, et al. (2001) Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol 70: 33-75.
- Kato M, Wynn RM, Chuang JL, et al. (2008) Structural basis for inactivation of the human pyruvate dehydrogenase complex by phosphorylation: Role of disordered phosphorylation loops. Structure 16: 1849-1859.
- Korotchkina LG, Patel MS (2001) Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem 276: 37223-37229.
- Korotchkina LG, Patel MS (2001) Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J Biol Chem 276: 5731-5738.
- Ferriero R, Iannuzzi C, Manco G, et al. (2015) Differential inhibition of PDKs by phenylbutyrate and enhancement of pyruvate dehydrogenase complex activity by combination with dichloroacetate. J Inherit Metab Dis 38: 895-904.
- Gudi R, Bowker-Kinley MM, Kedishvili NY, et al. (1995) Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem 270: 28989-28994.
- Ferriero R, Boutron A, Brivet M, et al. (2014) Phenylbutyrate increases pyruvate dehydrogenase complex activity in cells harboring a variety of defects. Ann Clin Transl Neurol 1: 462-470.
- Patel KP, O'Brien TW, Subramony SH, et al. (2012) The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol Genet Metab 105: 34-43.
- DeBrosse SD, Okajima K, Zhang S, et al. (2012) Spectrum of neurological and survival outcomes in pyruvate dehydrogenase complex (PDC) deficiency: Lack of correlation with genotype. Mol Genet Metab 107: 394-402.
- Quintana E, Gort L, Busquets C, et al. (2010) Mutational study in the PDHA1 gene of 40 patients suspected of pyruvate dehydrogenase complex deficiency. Clin Genet 77: 474-482.
- Naito E, Ito M, Yokota I, et al. (2002) Thiamine-responsive pyruvate dehydrogenase deficiency in two patients caused by a point mutation (F205L and L216F) within the thiamine pyrophosphate binding region. Biochim Biophys Acta 1588: 79-84.
- van Dongen S, Brown RM, Brown GK, et al. (2015) Thiamine-responsive and non-responsive patients with PDHC-E1 deficiency: A retrospective assessment. JIMD Rep 15: 13-27.
- Stacpoole PW, Gilbert LR, Neiberger RE, et al. (2008) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121: e1223-e1228.
- Ferriero R, Manco G, Lamantea E, et al. (2013) Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci Transl Med 5: 175ra31.
- Iannitti T, Palmieri B (2011) Clinical and experimental applications of sodium phenylbutyrate. Drugs RD 11: 227-249.
- Brockerhoff SE, Hurley JB, Janssen-Bienhold U, et al. (1995) A behavioral screen for isolating zebrafish mutants with visual system defects. Proc Natl Acad Sci U S A 92: 10545-10549.
- Brockerhoff SE, Dowling JE, Hurley JB (1998) Zebrafish retinal mutants. Vision Res 38: 1335-1339.
- Preiser JC, Moulart D, Vincent JL (1990) Dichloroacetate administration in the treatment of endotoxin shock. Circ Shock 30: 221-228.
- Ayala P, Montenegro J, Vivar R, et al. (2012) Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp Mol Pathol 92: 97-104.
- Srinivasan K, Sharma SS (2011) Sodium phenylbutyrate ameliorates focal cerebral ischemic/reperfusion injury associated with comorbid type 2 diabetes by reducing endoplasmic reticulum stress and DNA fragmentation. Behav Brain Res 225: 110-116.
- Qi X, Hosoi T, Okuma Y, et al. (2004) Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol 66: 899-908.
- Berg S, Serabe B, Aleksic A, et al. (2001) Pharmacokinetics and cerebrospinal fluid penetration of phenylacetate and phenylbutyrate in the nonhuman primate. Cancer Chemother Pharmacol 47: 385-390.
- Pliss L, Hausknecht KA, Stachowiak MK, et al. (2013) Cerebral developmental abnormalities in a mouse with systemic pyruvate dehydrogenase deficiency. PLoS One 8: e67473.
- Klejbor I, Mahmood S, Melka N, et al. (2020) Phenylbutyrate administration reduces changes in the cerebellar purkinje cells population in PDC-deficient mice. Acta Neurobiol Exp (Wars) 80: 305-321.
- Johnson MT, Mahmood S, Hyatt SL, et al. (2001) Inactivation of the murine pyruvate dehydrogenase (Pdha1) gene and its effect on early embryonic development. Mol Genet Metab 74: 293-302.
- Brown RM, Dahl HH, Brown GK (1990) Pyruvate dehydrogenase E1 alpha subunit genes in the mouse: Mapping and comparison with human homologs. Somat Cell Mol Genet 16: 487-492.
- Pliss L, Pentney RJ, Johnson MT, et al. (2004) Biochemical and structural brain alterations in female mice with cerebral pyruvate dehydrogenase deficiency. J Neurochem 91: 1082-1091.
- Pliss L, Mazurchuk R, Spernyak JA, et al. (2007) Brain MR imaging and proton MR spectroscopy in female mice with pyruvate dehydrogenase complex deficiency. Neurochem Res 32: 645-654.
- Morell P, Toews AD (1984) In vivo metabolism of oligodendroglial lipids. Oligodendroglia, Springer, Boston, MA.
- Connor JR, Menzies SL (1996) Relationship of iron to oligodendrocytes and myelination. GLIA 17: 83-93.
- Cammer W (1984) Carbonic anhydrase in oligodendrocytes and myelin in the central nervous system. Ann N Y Acad Sci 429: 494-497.
- Thorburne SK, Juurlink BH (1996) Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem 67: 1014-1022.
- Benarroch EE (2009) Oligodendrocytes: Susceptibility to injury and involvement in neurologic disease. Neurology 72: 1779-1785.
- Merrill JE, Ignarro LJ, Sherman MP, et al. (1993) Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol 151: 2132-2141.
- Soares-Fernandes JP, Teixeira-Gomes R, Cruz R, et al. (2008) Neonatal pyruvate dehydrogenase deficiency due to a R302H mutation in the PDHA1 gene: MRI findings. Pediatr Radiol 38: 559-562.
- Matute C, Alberdi E, Domercq M, et al. (2007) Excitotoxic damage to white matter. J Anat 210: 693-702.
- French HM, Reid M, Mamontov P, et al. (2009) Oxidative stress disrupts oligodendrocyte maturation. J Neurosci Res 87: 3076-3087.
- Volpe JJ, Kinney HC, Jensen FE, et al. (2011) The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int J Dev Neurosci 29: 423-440.
- Badaracco ME, Siri MV, Pasquini JM (2010) Oligodendrogenesis: The role of iron. Biofactors 36: 98-102.
- Burdo JR, Menzies SL, Simpson IA, et al. (2001) Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J Neurosci Res 66: 1198-1207.
- Lindboe CF, Torp SH (2002) Comparison of Ki-67 equivalent antibodies. J Clin Pathol 55: 467-471.
- Ohnishi Y, Iwatsuki K, Shinzawa K, et al. (2013) Adult olfactory sphere cells are a source of oligodendrocyte and Schwann cell progenitors. Stem Cell Res 11: 1178-1190.
- Menn B, Garcia-Verdugo JM, Yaschine C, et al. (2006) Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci 26: 7907-7918.
- Seri B, Garcia-Verdugo JM, Collado-Morente L, et al. (2004) Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J Comp Neurol 478: 359-378.
- Pavlu-Pereira H, Silva MJ, Florindo C, et al. (2020) Pyruvate dehydrogenase complex deficiency: Updating the clinical, metabolic and mutational landscapes in a cohort of Portuguese patients. Orphanet J Rare Dis 15: 298.
- Dasgupta S, Zhou Y, Jana M, et al. (2003) Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J Immunol 170: 3874-3882.
- Gardian G, Browne SE, Choi DK, et al. (2005) Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J Biol Chem 280: 556-563.
- Gupta N, Rutledge C (2019) Pyruvate dehydrogenase complex deficiency: An unusual cause of recurrent lactic acidosis in a paediatric critical care unit. J Crit Care Med (Targu Mures) 5: 71-75.
- Funfschilling U, Supplie LM, Mahad D, et al. (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485: 517-521.
- Della-Flora Nunes G, Mueller L, Silvestri N, et al. (2017) Acetyl-CoA production from pyruvate is not necessary for preservation of myelin. GLIA 65: 1626-1639.
Corresponding Author
Michal K Stachowiak and Ewa K Stachowiak, Department of Pathology and Anatomical Sciences, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, State University of New York, Buffalo, NY, 14203 USA; Western NY Stem Cell Center, Stereology Laboratory, University at Buffalo, State University of New York, Buffalo, NY, 14203 USA.
Copyright
© 2020 Freedman D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.