Kabuki Syndrome: A Rare Genetic Multisystem Disorder in Bangladesh
Introduction
Kabuki Syndrome (KS) is a rare multisystem genetic disorder. Patients present with unusual facial appearance with mental retardation along with other system involvement like cardiac, renal, neuropsychiatric disorder, hypodontia and post-natal growth retardation. Fundamental characteristics are called "Pentad of Niikawa" which includes dysmorphic face, skeletal abnormalities, dermatoglyphic abnormalities, mild to moderate mental deficit and post-natal growth retardation. Patients with KS are reported from different parts of globe.
Purpose of reporting the case of a Bangladeshi female child diagnosed as Kabuki syndrome with all her clinical features, laboratory tests and genetic report. This case highlights the existence of the syndrome with its complexities. Management of the patient is supportive by multidisciplinary team for better outcomes as there is no cure till now. Prevention approaches with promotion of growth and development of the affected children should be provided.
Detection of rare genetic case like Kabuki Syndrome (KS) is very important and challenging in developing countries like Bangladesh where scope of genetic test is limited. So we can adopt some physical signs as criteria for diagnosis and implication of a patient with KS.
Keywords
Kabuki Syndrome, Neuropsychiatry, Mental retardation, Hypodontia
Case Presentation
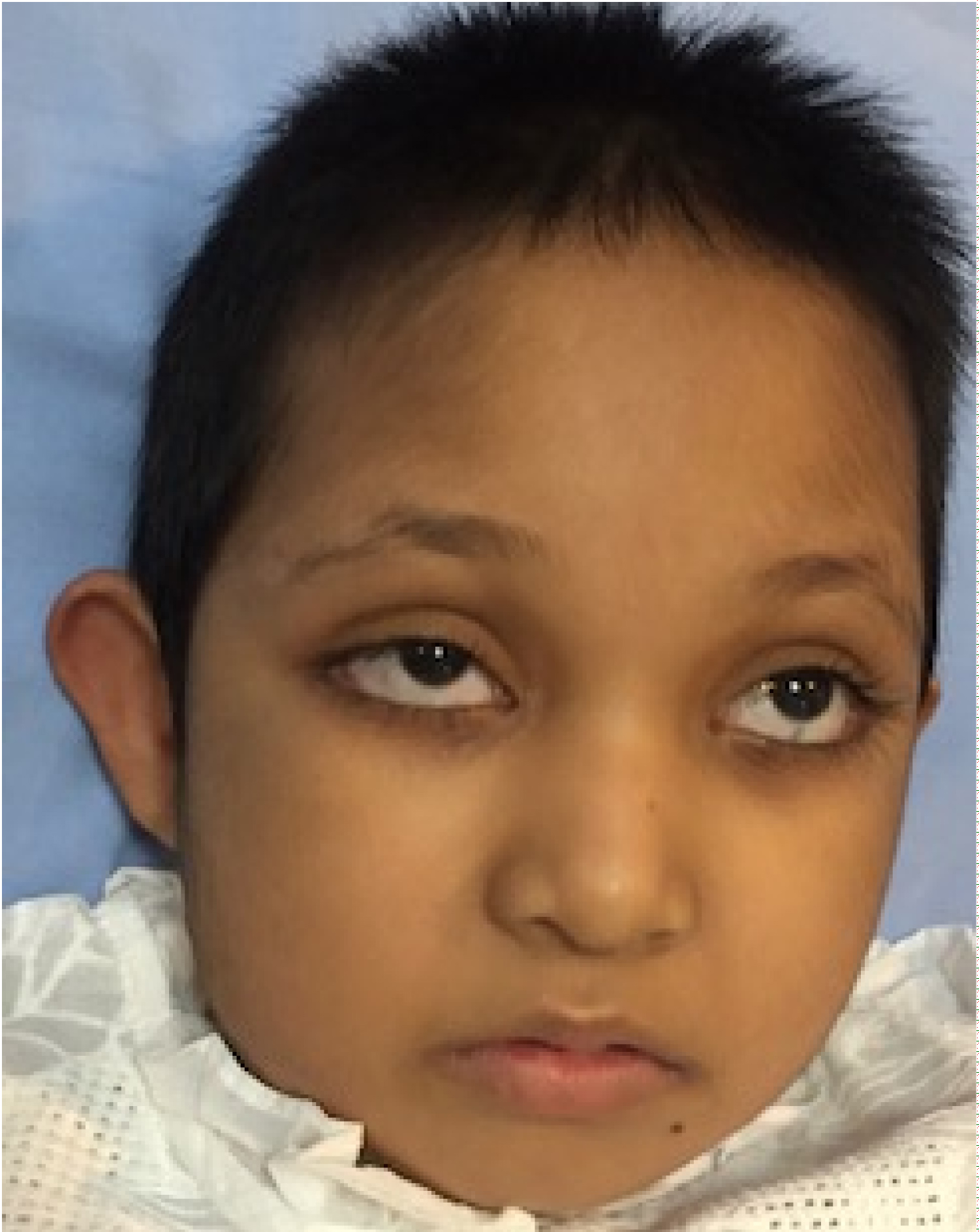
A 9-years 2-months-old Bangladeshi girl, 1st issue of non-consanguineous parents presented with complaints of fever, cough with a history of recurrent respiratory tract infection and was seen in our hospital. She was born of at term by LUCS weighed 2.6 kg. Antenatal history was unremarkable. She was born with congenital pneumonia, congenital heart disease and neonatal hyperbilirubinaemia and needed 2 weeks NICU management. She has delayed developmental milestones. She walked at 3 years of age, speech was delayed and not yet fluent. She has seizure with poor IQ (< 70). Family history was unremarkable. She had history of recurrent infection in early childhood. She is overweight (BMI-23.1 kg/m2, 97%), short statured (Height-118 cm, < 2 SD) and having microcephaly (OFC-47.5 cm, < 2 SD) with dysmorphic facies. Her clinical features comparing with major and minor criteria of Kabuki syndrome is given in Table 1 and Table 2 [1]. She was vitally stable, conscious with refractive error (myopia), hearing loss on right ear by audiogram, exaggerated jerks, extensor planter with trendelenburg gait. Her routine blood and urine test were normal with normal level of amino acids, organic acids and Fatty acids levels detected by Tandem Mass Spectrometry (TMS) Figure 1, Figure 2, Figure 3 and Figure 4.
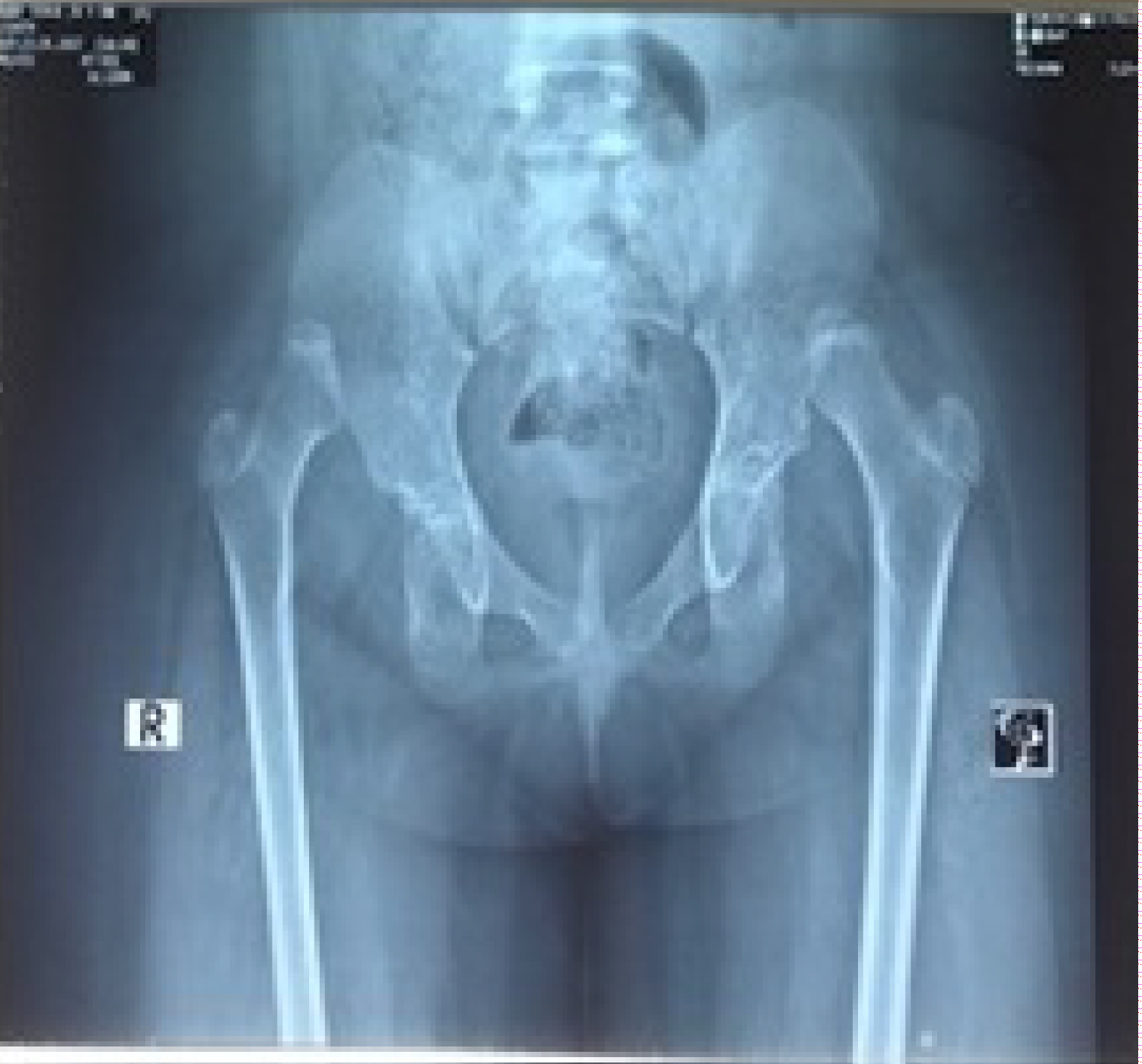
She has small sized and multicystic right kidney with normal left kidney detected by ultrasonography, echocardiography detected mild left pulminary artery origin stenosis, trival tricuspid regurgitation with abnormal interventricular septum motion, X-ray hip revealed congenital dislocation of both hip joints with under developed bones of pelvis, Electroencephalography detected focal epileptiform discharges over left frontal head region, Computed Tomography of brain was normal, karyotyping was shown 46 XX. Clinical exomesequencing revealed heterozygous mutation in KMT2D gene (Table 3). Therefore diagnosis was confirmed as Kabuki Syndrome-1.
Discussion
Kabuki syndrome is a rare genetic multisystem disorder. Initially it was thought to be specific to Japanese individuals only. One study was conducted over 62 patients in Japan and prevalence of KS was found in 1/32,000, all were sporadic, the sex ratio was even, there was no correlation with birth order, the consanguinity rate among the parents was not high and no incriminated agent was found that was taken by the mothers during early pregnancy [2]. However, there are reports of KS in a variety of ethnic groups including Northern European, Brazilian, Vietnamese, Filipino, East Indian, Arabic, Chinese, Mexican, and African [3]. The global incidence of KS is not known [4].
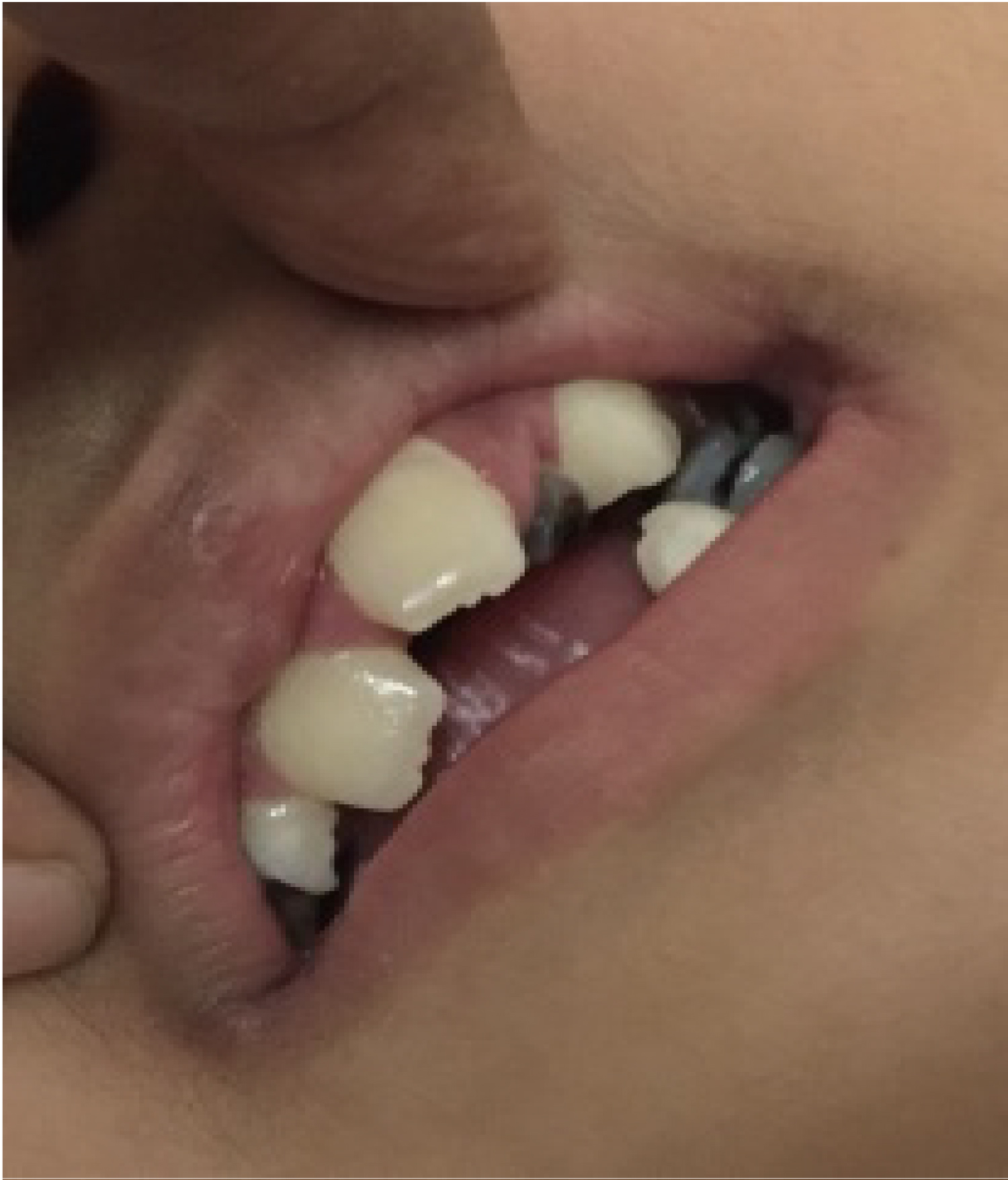
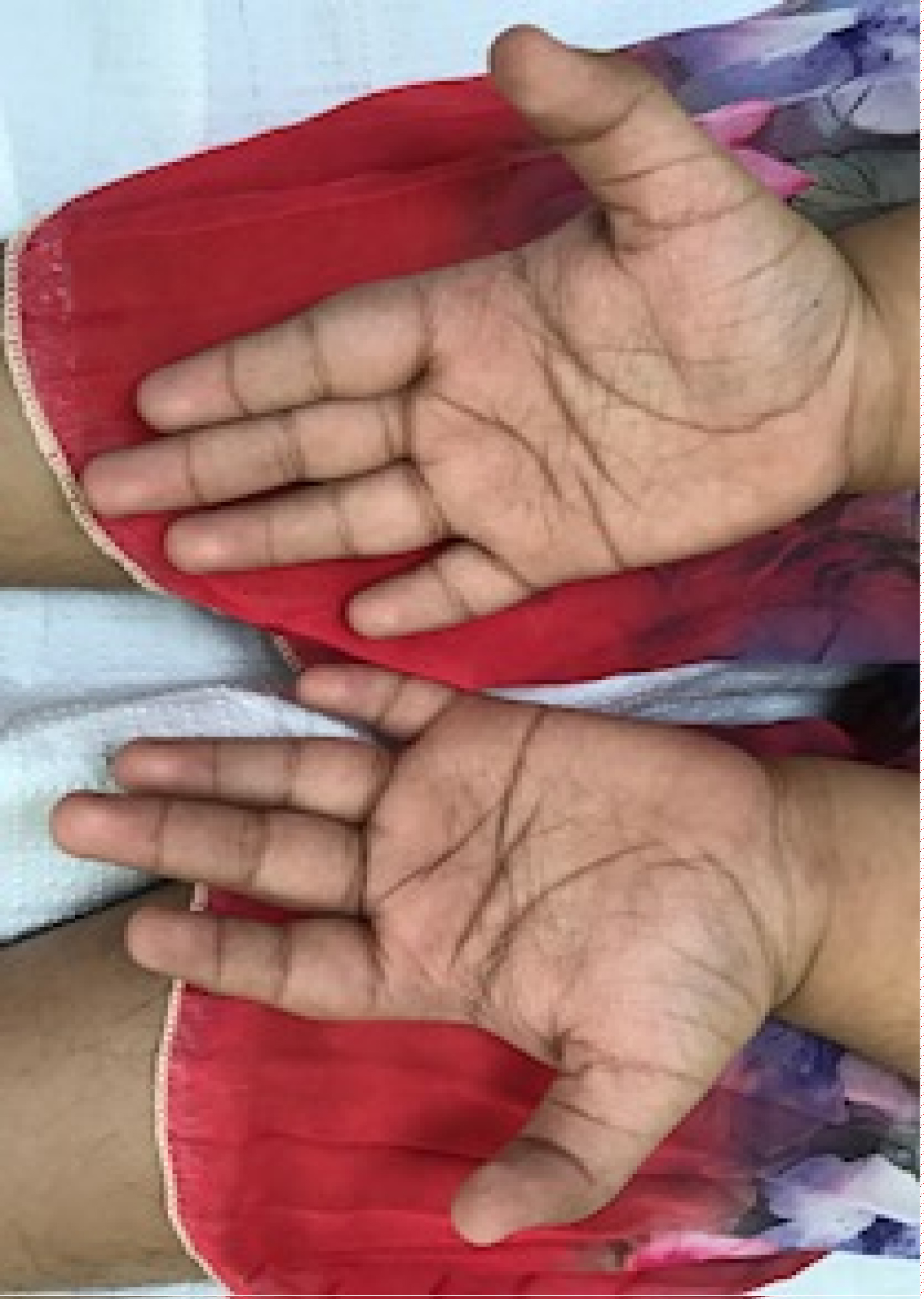
Clinical characteristics of Kabuki syndrome (KS) is characterized by typical facial features (long palpebral fissures with eversion of the lateral third of the lower eyelid; arched and broad eyebrows; short columella with depressed nasal tip; large, prominent, or cupped ears), minor skeletal anomalies, persistence of fetal fingertip pads, mild-to-moderate intellectual disability, and postnatal growth deficiency. Other findings may include: Congenital heart defects; genitourinary anomalies; cleft lip and/or palate; gastrointestinal anomalies including anal atresia, ptosis, and strabismus; and widely spaced teeth and hypodontia. Functional differences can include: Increased susceptibility to infections and autoimmune disorders, seizures, endocrinologic abnormalities (including isolated mature thelarche in females), feeding problems, and hearing loss [5].
Definitive diagnosis of KS can be made in an individual of any age with a history of infantile hypotonia, developmental delay and/or intellectual disability, and one or both of the following major criteria: 1) A pathogenic or likely pathogenic variant in KMT2D or KDM6A; and 2) Typical dysmorphic features (defined below) at some point of life. Typical dysmorphic features include long palpebral fissures with eversion of the lateral third of the lower eyelid and two or more of the following: a) Arched and broad eyebrows with the lateral third displaying notching or sparseness; b) Short columella with depressed nasal tip; c) Large, prominent or cupped ears; and d) Persistent fingertip pads [6,7]. Patient with KS may have multisystem involvement given in Table 4 [4,8].
In developing diagnostic criteria for KS, the expert panel performed a systematic review of information from the published literature on the features that are more specific for KS, particularly those features that are seen in individuals who have a known pathogenic variant in KMT2D. There is limited information on the range of clinical features seen in individuals who have a heterozygous or hemizygous pathogenic variant in KDM6A. While the typical facial gestalt and many of the other common features of KS, including mild humoral immunodeficiency, have been reported in these individuals as well, the frequency of such findings in larger cohorts of affected individuals is unknown [9-11]. Makrythanasis, et al. [12] identified 10 features found more commonly in individuals with pathogenic variants in KMT2D: Blue sclerae, arched eyebrows, broad nasal root, depressed nasal tip, large dysplastic ears, thin vermilion of the upper lip and thick vermilion of the lower lip, joint laxity, short stature, frequent infections, and intellectual disability. Characteristic dental abnormalities (absent lateral upper incisors, absent lower incisors and/or second premolars, abnormal 'flat head' screwdriver shape of the upper incisors) may be more common in those with a heterozygous pathogenic variant in KMT2D [13]. Furthermore, renal anomalies, premature thelarche in females, palatal anomalies and feeding problems are seen more commonly in those with a pathogenic variant in KMT2D compared with those who do not have a pathogenic variant in KMT2D [14-16]. Makrythanasis, et al. [12] developed a phenotypic scoring system with the purpose of determining which individuals were more likely to have a pathogenic variant in KMT2D. Two small validation studies of the scoring system were then published by Paděrová, et al. [17] and Paderova, et al. [18]. In both studies the number of individuals included was small and overlapping.
Using the clinical scoring system reported by Makrythanasis, et al. [12] it was found that individuals with a score above 6.0, had a pathogenic variant in KMT2D and no individual with a score below 5.0 was found to have a pathogenic variant in KMT2D. But limitation of the phenotypic scoring system is that it is not applicable to the neonate. Therefore, in order to make a definitive or probable clinical diagnosis of KS, long palpebral fissures with eversion of the lateral third of the lower eyelid at some point of life is required. The authors applied the proposed diagnostic criteria to three individuals with mosaic KS reported in the literature [19]. Each reported patient had a mosaic heterozygous pathogenic variant in KMT2D and was reported to have mild clinical features of KS. Using the proposed clinical diagnostic criteria (ignoring the molecular confirmation), two out of three of these reported individuals would have been given a definitive clinical diagnosis of KS [6] Table 5.
According to the clinical scoring system reported by Makrythanasis, et al. [12] our reported case scores above 8.0 and clinically diagnosed as KS which was confirmed of heterogenous mutation of KMT2D gene by clinical exome sequencing. Her management continued with antiepileptic drug for seizure control with speech therapy, occupational and physiotherapy. Genetic counseling was done. She is attending inclusive school and her activity of daily living mostly independent. Appearance of her parents and younger sib are normal and her sib studies in main stream school. Family screening was advised but not yet done.
Genetic basis of Kabuki syndrome is heterozygous mutations in the KMT2D gene (also known as MLL2) inherited in an autosomal dominant pattern, known as Kabuki syndrome type-1 and is about 55-80% of total case. Another mutation in the KDM6A gene inherited in an X-linked dominant pattern, known as Kabuki syndrome type-2 and it is about 2-6% [15,20,21]. Some people with Kabuki syndrome have no identified KMT2D or KDM6A gene mutation. The cause of the disorder in these individuals is unknown. Most cases of Kabuki syndrome are not inherited from a parent and result from a new mutation in one of these genes (in people with no history of Kabuki syndrome) [8].
Management of the patients with KS is supportive by the multidisciplinary team for better outcomes as there is no cure till now. It requires an individualized, multidisciplinary and coordinated plan of a team of specialists who will provide treatment and support of the patient as required. It can include a paediatrician, developmental paediatrician, cardiologist, orthopedic surgeon, dental surgeon, endocrinologist, speech/language therapist, developmental and occupational therapist, geneticist and an immunologist, although need for these subspecialists depend upon the patient's phenotype [22].
In conclusion, presentation of KS is variable as per reviewing of different journal and articles. They may present in various way like hypotonia, recurrent infections, growth retardation/obesity, hearing or visual problem, neuropsychiatric disorders or language problem, epilepsy along with characteristic facial findings. It is very important to be focused on the existence of the syndrome and its complexity and should be able to diagnose clinically by using the proposed clinical diagnostic criteria (ignoring the molecular confirmation) and early management by multidisciplinary team depends on patient phenotype as there is no specific treatment available yet. Further research is recommended for specific management in future.
References
- Sattur A, Deshmukh PK, Abrahim L, et al. (2014) Kabuki make-up syndrome: A case report with electromyographic study. J Clin Diagn Res 8: 03-06.
- Niikawa N, Kuroki Y, Kajii T, et al. (1988) Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 62 patients. Am J Med Genet 31: 565-589.
- Adam MP, Hudgins L (2005) Kabuki syndrome: A Review. Clin Genet 67: 209-219.
- Cheon CK, Ko JM (2015) Kabuki syndrome: Clinical and molecular characteristics. Clin Exp Pediatr 58: 317-324.
- Adam MP, Hudgins L, Hannnibal M (2019) Kabuki syndrome Synonyms: Kabuki Make-Up Syndrome, Niikawa-Kuroki Syndrome. GeneReviews®. Initial Posting: September 1, 2011; Last Revision: October 21, 2019.
- Adam MP, Banka S, Bjornsson HT, et al. (2019) Kabuki syndrome: International consensus diagnostic criteria. J Med Genet 56: 89-95.
- OMIM Entry - # 147920 - KABUKI SYNDROME 1; KABUK1.
- Matsumoto N, Niikawa N (2003) Kabuki make-up syndrome: A review. Am J Med Genet C Semin Med Genet 117: 57-65.
- Bogershausen N, Gatinois V, Riehmer V, et al. (2016) Mutation update for kabuki syndrome genes KMT2D and KDM6A and further delineation of x-linked kabuki syndrome subtype 2. Hum Mutat 37: 847-864.
- Frans G, Meyts I, Devriendt K, et al. (2016) Mild humoral immunodeficiency in a patient with X-linked Kabuki syndrome. Am J Med Genet A170: 801-803.
- Margot H, Genevieve D, Gatinois V, et al. (2016) Typical facial gestalt in X-linked Kabuki syndrome. Am J Med Genet A 170: 3363-3364.
- Makrythanasis P, van Bon BW, Steehouwer M, et al. (2013) MLL2 mutation detection in 86 patients with Kabuki syndrome: A Genotype-phenotype study. Clin Genet 84: 539-545.
- Porntaveetus T, Abid MF, Theerapanon T, et al. (2018) Expanding the Oro-Dental and Mutational Spectra of Kabuki Syndrome and Expression of KMT2D and KDM6A in Human Tooth Germs. Int J Biol Sic 14: 381-389.
- Hannibal MC, Buckingham KJ, Ng SB, et al. (2011) Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A 155: 1511-1516.
- Li Y, Bogershausen N, Alanay Y, et al. (2011) A mutation screen in patients with Kabuki syndrome. Hum Genet 130: 715-724.
- Paulussen AD, Stegmann AP, Blok MJ, et al. (2011) MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat 32: E2018-E2025.
- Paderova J, Holubova A, Simandlova M, et al. (2016) Molecular genetic analysis in 14 Czech Kabuki syndrome patients is confirming the utility of phenotypic scoring. Clin Genet 90: 230-237.
- Paderova J, Drabova J, Holubova A, et al. (2018) Under the mask of Kabuki syndrome: Elucidation of genetic-and phenotypic heterogeneity in patients with Kabuki-like phenotype. Eur J Med Genet 61: 315-321.
- Lepri FR, Cocciadiferro D, Augello B, et al. (2017) Clinical and neurobehavioral features of three novel kabuki syndrome patients with mosaic kmt2d mutations and a review of literature. Int J Mol Sci 19: 82.
- ghr.nlm.nih.gov › condition › kabuki-syndrome
- Banka S, Veeramachaneni R, Reardon W, et al. (2012) How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet 20: 381-388.
- http://www.kennedykrieger.org/sites/default/files/library/documents/patient-care/conditions/kabukisyndrome/ kabuki-syndrome-services-at-kennedy-krieger-institute-7-7-17_0.pdf.
Corresponding Author
Zeena Salwa, Consultant, Paediatrics & Child Development Center, Square Hospitals Ltd, Dhaka, Bangladesh, Tel: +8801715059815.
Copyright
© 2020 Salwa Z, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.