Sacral Chordoma: A Case Report
Abstract
Sacral chordoma is a tumor that corresponds to 1 to 4% of all malignant bone tumors in the axial skeleton; It consists of a silent and slow-growing mass that requires radical treatment due to its high potential for local and metastatic invasion, its surgical treatment and management decision constitutes a challenge for the surgeon who faces it given the morbidity and affectation in the quality of life of the patient who in most cases is asymptomatic; We present the case of a 75-year-old patient who debuts with a giant sacral mass that generates very scarce symptoms in the context of an otherwise healthy patient, an analysis of the case is carried out regarding its clinical and diagnostic aspects.
Keywords
Chordoma, Sacral chordoma, Sacrectomy, Spine surgery, Orthopaedic spine surgery, Sacral tumor
Introduction
Sacral chordoma is the most common malignant primary tumor of the axial skeleton, it corresponds to an asymptomatic, slow growing, but aggressive lesion with the potential for local and metastatic invasion. Its diagnosis is made by images and mainly pathological study accompanied by immunohistochemistry. The gold standard for treatment corresponds to a challenge for the treating surgeon given its high risk of associated morbidity and mortality and especially secondary neurological injury; for this reason, its early diagnosis and suspicion are important, as well as the discussion regarding its management and evaluation of the risk benefit for the patient, their doctor and their family.
Case Presentation
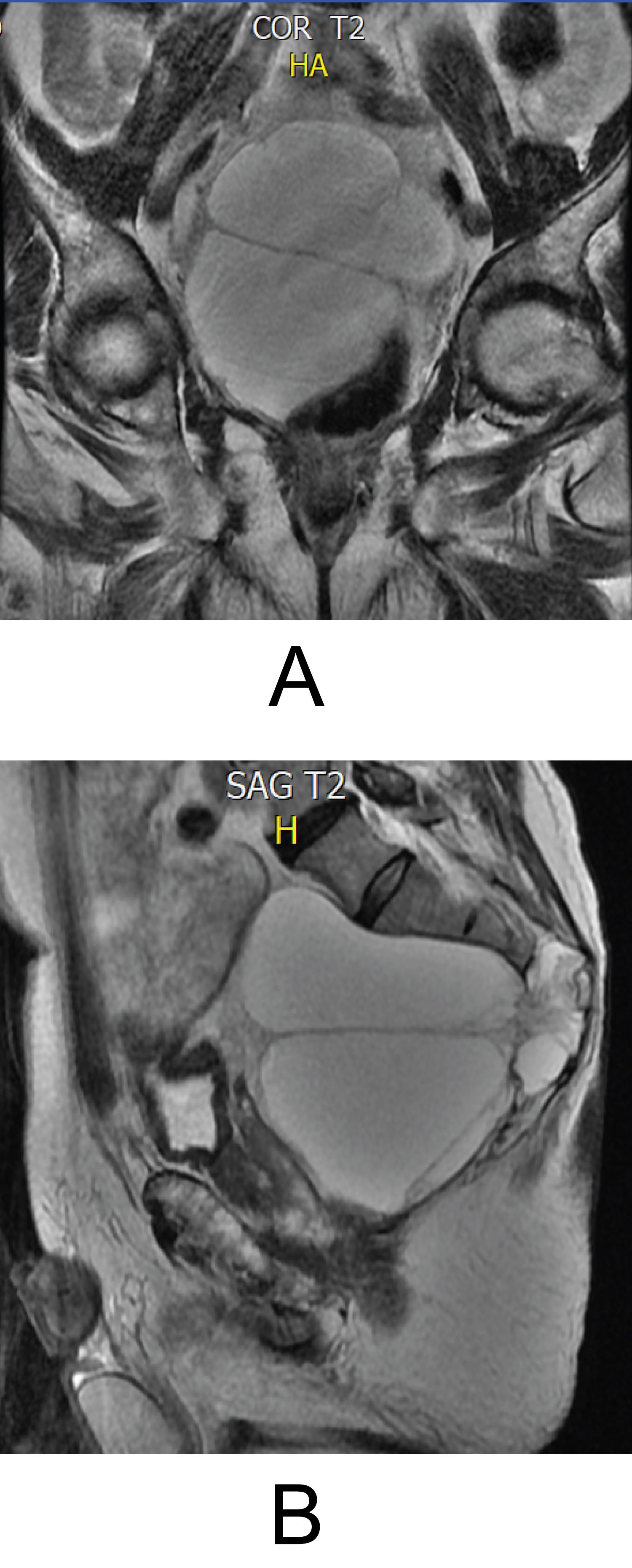
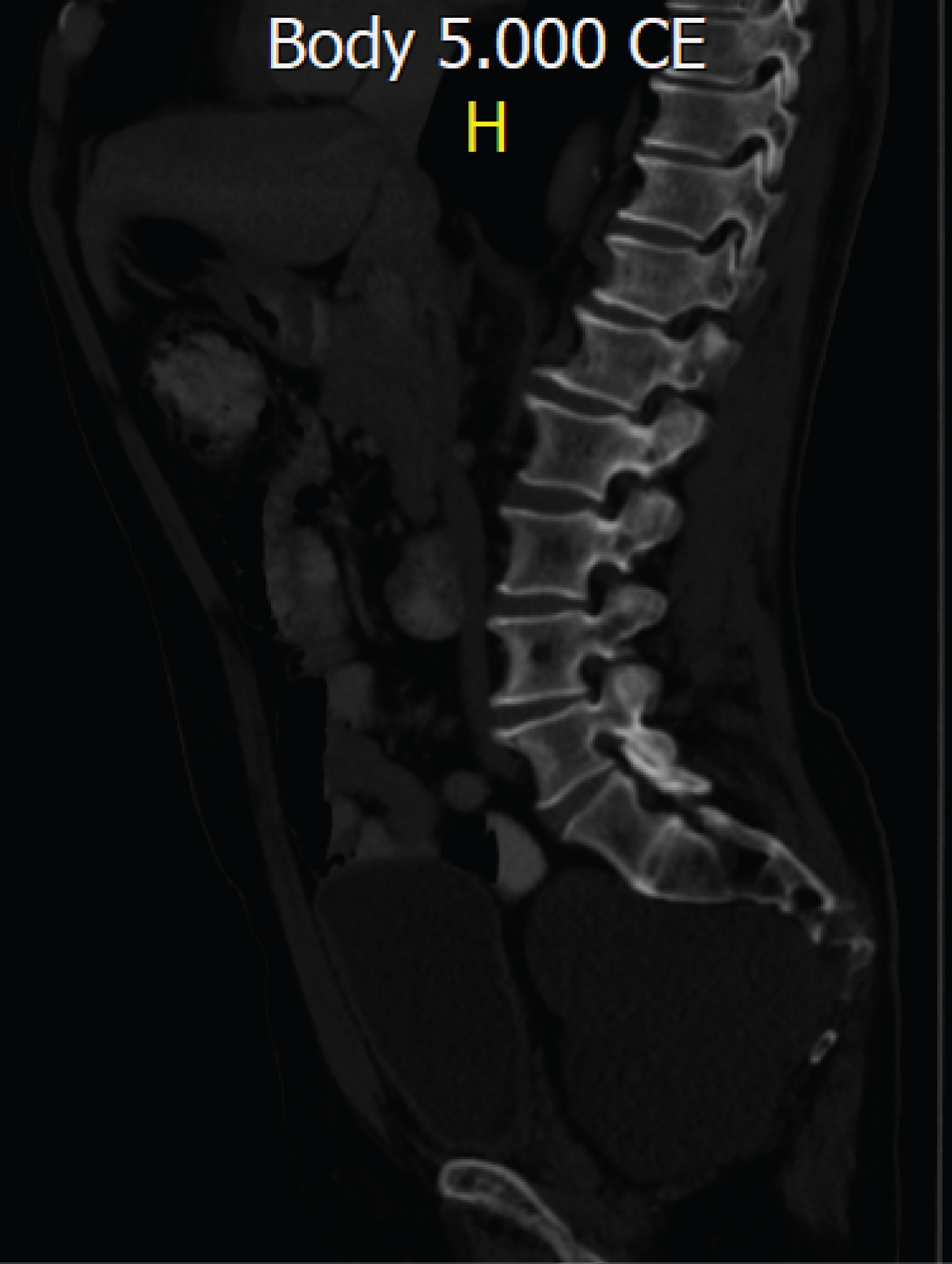
A 75-year-old patient with a 2-year evolution consisting of right groin pain, flexion of the right hip limitation and discomfort when walking and climbing stairs, as the only important antecedent refers to transurethral resection of the prostate due to hyperplasia. The patient is initially evaluated by general orthopedics who refer a subspecialist in spine surgery to rule out pathology in the hip and lumbar spine with X-rays, to the physical examination of the patient didn't present neurological deficit of the lower limbs or signs of neurological targeting and/or spinal or root compression, MRI of the lumbosacral spine is requested, which reports a lesion that involves the sacrum at the level of S3, S4 and S5 with growth towards the presacral space, displacing pelvic structures anteriorly and involvement of the spinal canal and foramina of S3 and S4 (Figure 1A and Figure 1B), a TRUCUT biopsy was performed which reports an infiltrating sacrococcygeal lesion with musculoskeletal and bone connective tissue with myxoid change suggestive of chordoma, positive immunohistochemical report for: CKAE1AE3, CAM5,2, CK19, EMA (WEAK), S100 (FOCAL) and negative for: CK7, PGFA, CDX2, TTF1, PSA, CEA with a cell proliferation index of 8%. During the follow-up, it was also decided to perform a thoracic-abdominal-pelvic CT scan to assess the presence of additional metastatic lesions (negative) and delimit osseous sacral involvement (Figure 2).
Given the diagnosis Chordoma dependent on the last segments of the sacrum, it was decided to take the patient to the institutional surgical medical board to define definitive management, after a comprehensive review of the literature, it was decided together with the patient to carry out expectant management of the lesion given the scarce symptoms, the age of the patient, the morbidity of the surgery, the high risk of permanent neurological injury to the sacral roots during sacrectomy with secondary loss of sphincter control and probable compromise of gait, and the high risk of recurrence of the injury after surgery, resection of the tumor.
Discussion
It is vitally important to recognize the existence of the sacral chordoma as the first most prevalent primary tumor of the spine and the importance of its management and approach in continuous change. Chordoma corresponds to 1 to 4% of malignant bone tumors, it is a slow-growing lesion, generally asymptomatic and with a high recurrence [1,2], this tumor has a bimodal presentation, being prevalent between 50 to 60 years of age and less than 5% of cases in children and adolescents [1,3]; its location is almost always in the spine, where it exhibits aggressive behavior given its local invasive potential, being capable of generating metastases that are generally found in the lungs, liver and extra-axial bone [1,2].
The origin of the chordoma is not clear, it is proposed that it comes from cells of the notochord leaving some vestiges in the bones of the spine, the base of the skull and the nucleus pulposus of the intervertebral discs (NOT known to be the cause of the tumor) [1,2]; The first description of chordoma clinically was made in 1857 by Virchow, who histologically describes a lesion with unique intracellular "bubble-like" vacuoles, later in 1985, Ribbert associated the lesion with notochord cells in 0.4 to 2% of the autopsies performed finding the tumor as a mass of less than 1 centimeter located behind the clivus [2,3].
To date, no environmental, dietary or lifestyle risk factors have been correlated with the appearance of the tumor, however, there is a found relationship with the mutation in the 6q27 locus of the "T" Gene (transcription factor) that encodes the protein "Brachyuria" generating an abnormal notochord without being the direct etiology of the tumor (probability less than 2 in a million). There is concomitant familial chordoma conditioned by an extra copy of the brachyuria gene; this form of presentation is much more infrequent and tends to occur in patients with tuberous sclerosis [1,2,4]. The hypothesis of the origin in the notochord is supported by 3 conditions: similar morphology between these remnants and the histopathology, the site of the vestiges of the notochord corresponds to the distribution of chordomas and the similar immunophenotype [3].
During the histopathological study of the lesion, the chordoma appears as a grayish to dark lobulated mass with calcifications, gelatinous and vacuolated cells called "physaliphores" (with bubbles or vacuoles), which are surrounded by myxoid matrix [2,5]. The types of chordoma according to their histology are: conventional (or classic - the most common), poorly differentiated (aggressive and fast-growing type related to the loss of the INI-1 gene in pediatric patients and young adults), Dedifferentiated (or sarcomatous - the most aggressive also associated with the loss of the INI-1 gene in pediatric patients) and Chondroid (presence of the brachyuria gene, which is expressed in almost all conventional chordomas and gives the difference with chondrosarcoma) [1,4].
Clinically, chordoma is a generally asymptomatic pathology, however depending on its location it can generate symptoms related to its location: Skull base with cranial nerve palsy, endocrinopathy (according to size), epistaxis and intracranial hemorrhage; mobile spine and sacrum with deep localized pain, radiculopathy (location dependent), compromise of bladder or intestinal control and if it is cervical even obstruction of the airway, dysphagia and/or oropharyngeal mass [3]. the diagnosis of chordoma is generally late, in which case 65% of patients already have metastases (usually its sacral locationis not evaluated in most of the resonance protocols) [1,3,5].
Radiography, tomography and MRI are useful in the initial approach, usually a midline lesion with lytic characteristics and epicenter in the vertebral body dependent on soft tissue is shown; calcifications and/or bone expansion. Once the image approach is done, the definitive diagnosis is stablished by biopsy and immunohistochemistry with markers such as S100, epithelial markers and cytokeratins (versus chondrosarcoma) and of course to the aforementioned brachyuria; The latter has shown a sensitivity of 98% and a specificity of 100% in chordoma samples positive for cytokeratins [3,5].
The treatment of chordoma corresponds to an interdisciplinary approach, the gold standard to date and supported by many case series consists of bloc resection of the lesion with wide margins (sacrectomy) and postoperative radiotherapy [3,4,6], the complete resection of the tumor is the most important prognostic factor. Sacrectomy is a procedure with high morbidity mainly due to the risk of resection of the roots of S1, in which case ambulation, sphincter function and sexual impotence is compromised; even in the best ofcases, keeping the two roots of S2 there is a 50% risk of losing bladder and bowel control. The second major risk consists of rupture of the capsule during resection with the possibility of tumor seeding and secondary recurrence [3], Kaiser, et al. identify that the tumor can have a local recurrence twice as high if a resection with violation of margins is performed [5]. Patients with chordoma without treatment have shown an average survival of 18 months given the local progression and the risk of metastasis. The most important factor of mortality corresponds to local recurrence, Radical versus subtotal resection in sacral tumor presents a recurrence rate of 2.27 years compared with 8 months respectively [5], Also the size of the mass has shown risk of recurrence being 46% for tumors larger than 10 cm. and 17% for minors [6,7], the involvement of adjacent structures reports a correlation of 90% in case of sacroiliac joint involvement and 85.7% in invasion of the piriformis and gluteus medius (9), the 5-year evidence of postoperative metastases corresponds to 40% and the 5-year postoperative survival corresponds to 45 to 70% [6].
The treatment also includes adjuvant radiation, conventional radiotherapy (RT) (local tumor control at 5 years of 10 to 40%) and proton radiotherapy (5-year survival of 73%). Regarding chemotherapy, studies point to its use as adjuvant therapy, however, so far, the evidence is limited, the studies few and the follow-ups very short [3].
The average half-life in general is 67.6% at 5 years, 39.9% at 10 years and 13.1% at 20 years, being mainly affected by local invasion rather than metastasis [2,3,5]. the current approach and the future for this tumor consists of better understanding of its molecular basis, improving the management for local recurrence, optimizing adjuvant radiotherapy and expanding studies on the brachyuria gene to achieve senescence of tumor cells [4].
Conclusion
Sacral chordoma is a rare tumor, it consists of a slowly growing but aggressive and locally invasive mass, with a high recurrence rate. The gold standard management consists of complete resection and adjuvant radiotherapy, however, this surgical management implies high morbidity and a severe compromise of the quality of life of the patient, who at first most of the time has presented as asymptomatic.
References
- (2018) Understanding Chordoma-Chordoma Foundation. (5th edn).
- Bresson D, Froelich SC, George B, et al. (2015) Chordomas: A Review. Neurosurg Clin N Am 26: 437-452.
- Coumans J, Ferreira M, Kahle KT, et al. (2012) Chordoma: Current concepts, management, and future directions. Lancet Oncol 13: e69-e76.
- Awad OA, Gokaslan ZL, Hsu W, et al. (2011) Generation of chordoma cell line JHC7 and the identification of Brachyury as a novel molecular target. J Neurosurg 115: 760-769.
- Gompel JJ, Janus JR (2015) Chordoma and chondrosarcoma. Otolaryngol Clin North Am 48: 501-514.
- Briggs TW, Hanna SA, Kayani B, et al. (2014) A review of the surgical management of sacral chordoma. Eur J Surg Oncol 40: 1412-1420.
- Atalar AC, Eralp L, Ozger H, et al. (2010) Surgical management of sacral chordoma. Acta Orthop Belg 76: 243-253.
Corresponding Author
Solángel Rodríguez M, MD, Orthopedic Surgeon, Pontificial Javeriana University, Bogotá, Colombia
Copyright
© 2021 Solángel RM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.