Development of Hodgkin's Lymphoma-Like Post-Transplant Lymphoproliferative Disorder after Tandem Autologous Peripheral Blood Stem Cell Transplantation for Multiple Myeloma
Abstract
Post-Transplant Lymphoproliferative Disorders (PTLDs) are serious complication after solid organ or Hematopoietic Stem Cell Transplantation (HSCT). Hodgkin's Lymphoma (HL)-like PTLD is grouped with HL type PTLD in the World Health Organization. HL type PTLD is the rarest form of PTLD. Furthermore, the development of HL type PTLD after autologous HSCT is extremely rare. In the present study, we report a case of HL-like PTLD after tandem autologous peripheral blood stem cell transplantation for multiple myeloma. A 69-years-old Japanese male was admitted to our hospital because of the detection of an Immunoglobulin (Ig)G elevation in July 2005. The serum IgG level was 7512 mg/dl. Analysis using serum electrophoresis and immunoelectrophoresis revealed IgG λ monoclonal protein. Stage II multiple myeloma according to the International Staging System was diagnosed. After the forth cycle of VAD chemotherapy, peripheral blood stem cells were harvested and the patient underwent tandem autologous Peripheral Blood Stem Cell Transplantation (PBSCT), with high-dose melphalan as conditioning regimen. 13 months after tandem autologous HSCT, he presented with high fever and swelling of the left cervical lymph nodes. A biopsy showed involvement of malignant lymphoma, which was composed of large Reed-Stemberg (RS)-like cells and small to medium lymphocytes. Morphological picture suggested HL type PTLD. Immunohistochemical study disclosed that the RS-like cells were positive for CD20, CD30 and negative for CD15. CD20-positive cells were positive for EBV-Encoded Early RNA (EBER). These findings suggested HL-like PTLD rather than HL type PTLD. Despite treatment with chemotherapy, the patient died of multi-organ failure on the 18th day after admission.
Keywords
Hodgkin's lymphoma, Post-Transplant Lymphoproliferative Disorders, Autologous Hematopoietic Stem Cell Transplantation
Introduction
Post-Transplant Lymphoproliferative Disorders (PTLDs) are lymphoid or plasmacytic proliferations that occur in immunosuppressed recipients following solid organ or Hematopoietic Stem Cell Transplantation (HSCT). In both solid organ transplantation and HSCT, more than 80% cases are associated with Epstein-Barr virus (EBV). For solid organ transplantation recipients, the highest incidence of PTLD is after small bowel transplantation (20%), followed by lung (10%), heart (6%), liver (2.8%), and renal (2.3%) transplantation. The incidence of PTLD in allogenic HSCT has been reported between 0.5% and 2.5% [1]. Compared to the allogenic setting, the development of PTLD is very rare complication in autologous HSCT [2].
The World Health Organization (WHO) classifies PTLD into four categories: early lesions, polymorphic PTLD, monomorphic PTLD, and Hodgkin's Lymphoma (HL) type PTLD that lists both classical HL type PTLD and HL-like PTLD. HL type PTLD is the rarest form of PTLD [3]. HL type PTLD after autologous HSCT is extremely rare, only four cases have been reported [4-6], and HL-like PTLD after autologous HSCT has not been reported. We report here a case of HL-like PTLD that developed following tandem autologous HSCT for multiple myeloma.
Case Report
A 69-years-old Japanese male without a noteworthy past history was admitted to our hospital because of the detection of an IgG elevation in July 2005. The serum IgG level was 7512 mg/dl. Analysis using serum electrophoresis and immunoelectrophoresis revealed IgGλ monoclonal protein. Bone marrow aspiration disclosed 20.8% myeloma cells. Laboratory test results included a hemoglobin level of 8.8 g/dl, a serum β-2 microgloblin level of 3.1 mg/l, and a serum albumin level of 3.5 g/dl. Stage II multiple myeloma according to the International Staging System was diagnosed. The patient initially received VAD chemotherapy (0.4 mg/m2 of vincristine on days 1-4, 9 mg/m2 doxorubicin on days 1-4, 40 mg dexamethasone on days 1-4, 9-12, 17-20). After the forth cycle of VAD chemotherapy, he achieved Very Good Partial Response (VGPR) according to International uniform response criteria for multiple myeloma and peripheral blood stem cells were harvested. For mobilization 4000 mg/m2 cyclophosphamide plus G-CSF was administered. In February 2006, the patient underwent autologous PBSCT, with high-dose melphalan (200 mg/m2) as conditioning regimen and obtained VGPR. In July 2006, he received a second autologous PBSCT under same conditioning regimen. After second transplantation, he remained in VGPR without maintenance therapy.
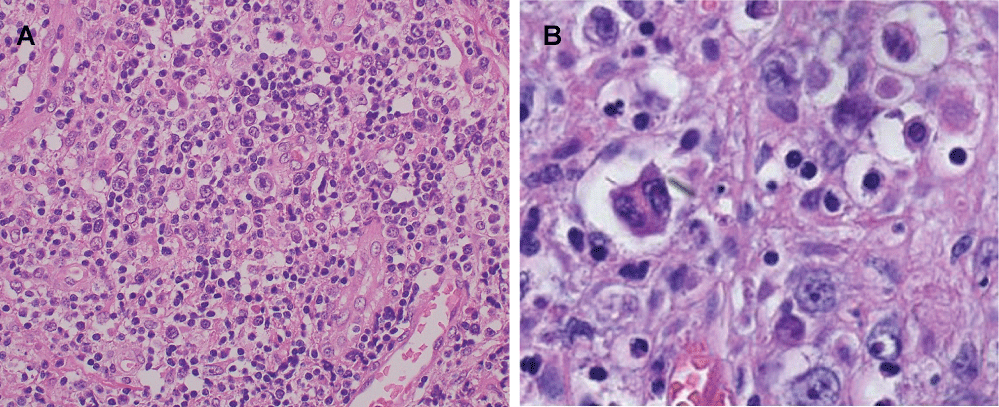
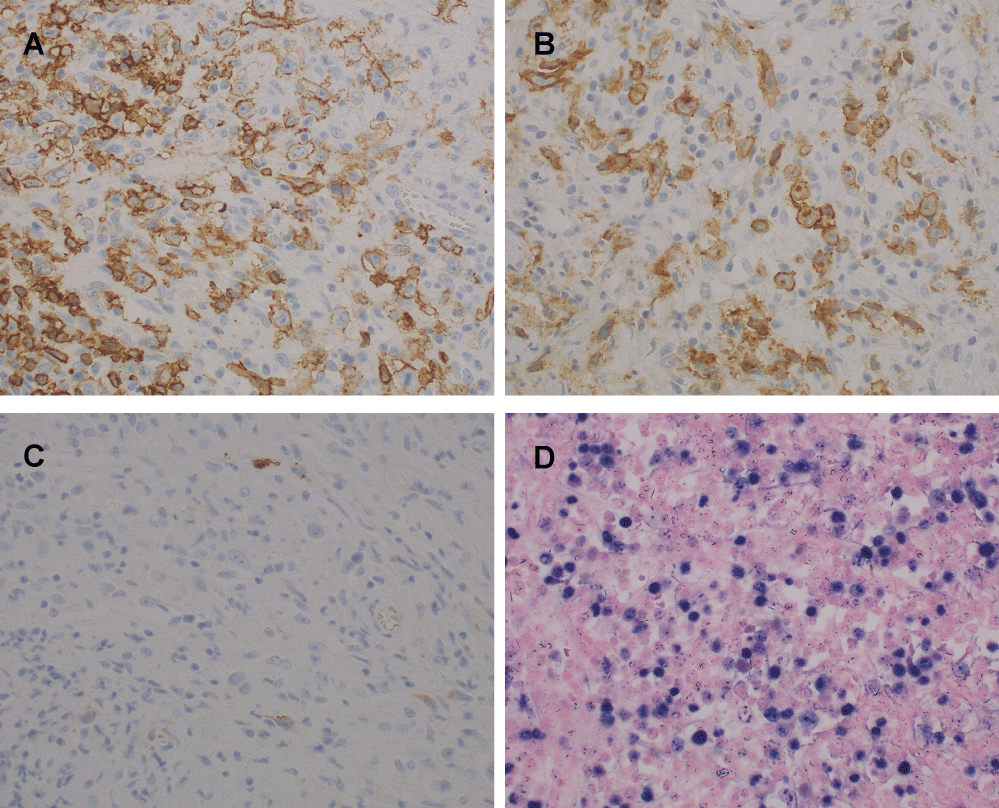
In October 2007, the patient was admitted to hospital again due to high fever and swelling of the left cervical lymph nodes. On physical examination, he had several lymph nodes in his left neck. Laboratory data were as follows: white cell count 4.9 × 109/l, hemoglobin 10.0 g/dl, platelet count 192 × 109/l, total bilirubin 0.29 mg/dl, lactate dehydrogenase 283 IU/L, and soluble Interloikin-2 Receptor (sIL-2R) 3890 U/ml (Table 1). Immunoelectrophoresis showed no increase in serum monoclonal Ig. Computed Tomography (CT) revealed swelling of the tonsil and enlarged cervical, supraclavicular, mediastinal, and paraaortic lymph nodes and several low density areas in the spleen. Serum titers of the antibodies against EBV were as follows: Virus Capsid Antigen (VCA) IgG, x1280; VCA IgM, < x10; Early Antigen, Diffuse Type and Restricted Type Antibody (EA-DR) IgG, x20; EBV Nuclear Antigen (EBNA), < x10 (Table 1). Cervical lymph node biopsy was performed and histologic examination of the lymph node showed that normal lymph node architecture was not preserved and binucleated Reed-Stemberg (RS) cells with eosinophilic nucleoli and atypical large lymphocytes with single nuclei were scattered among small to medium-sized lymphocytes (Figure 1). Immunohistochemically, these atypical cells were positive for CD20, CD30 and negative for CD15, CD3, CD10, ALK, EMA. EBV-Encoded Early RNA (EBER) was detected in both the atypical large lymphocytes and the subset of small background lymphocytes by in situ hybridization (Figure 2). These findings were consistent with a diagnosis of HL-like PTLD.
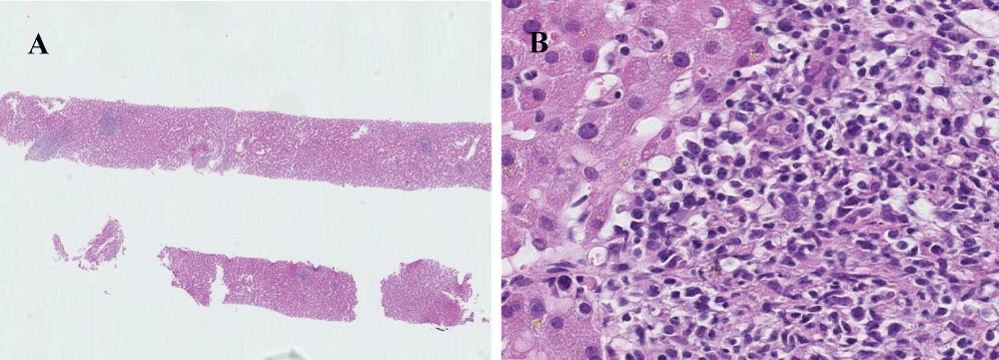
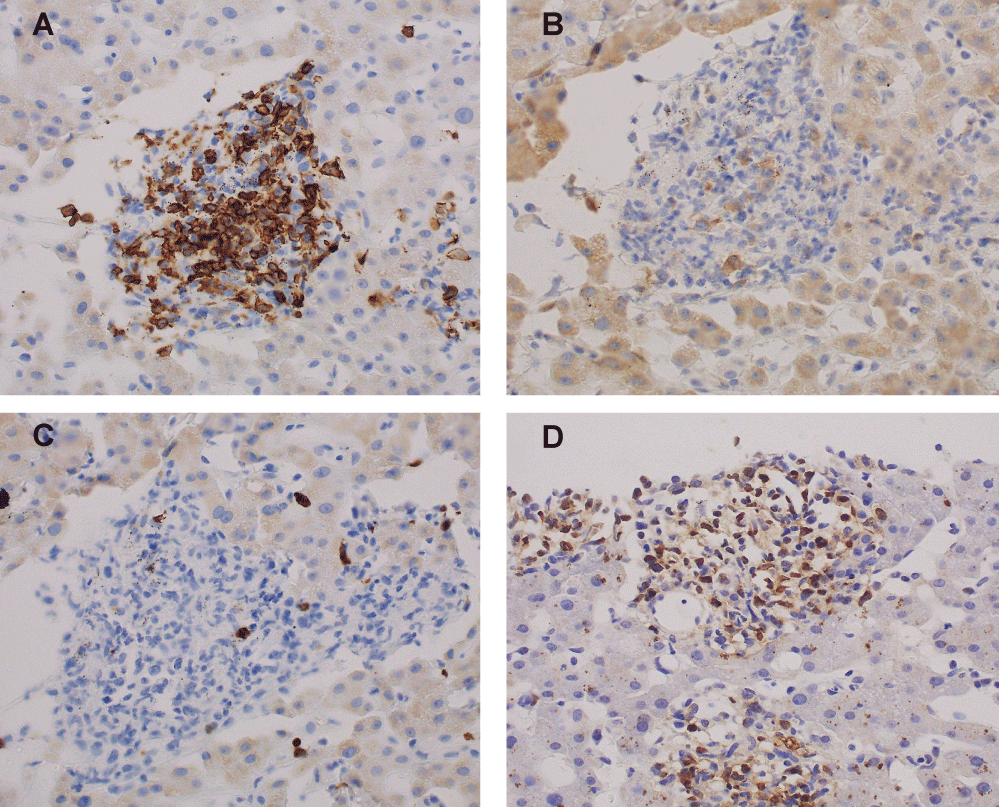
On the fourth day after hospitalization, liver dysfunction developed. Needle liver biopsy was performed. On histological examination of the liver, portal infiltration of atypical large cells with typical small to medium-sized lymphocyte background was observed (Figure 3). Immunohistochemical analyses demonstrated that large atypical cells were positive for CD20, CD30, and EBER and negative for CD15, CD3 (Figure 4). In consideration of a liver damage, the patient was treated with mini-MEAM therapy except etoposide (60 mg/m2 of ranimustine on day 1, 100 mg/m2 of cytarabine on days 2-5, 30 mg/m2 of melphalan on day 6), because these drugs are less likely to cause a liver damage. However, the chemotherapy was not effective. A condition of the patient and liver function tests worsened rapidly (Table 2). In addition, he developed respiratory and renal failure. The patient died of multi-organ failure on the 18th day after admission.
Discussion
In this report we describe the occurrence of HL-like PTLD as a secondary neoplasm after tandem autologous HSCT for multiple myeloma. Autologous HSCT is the standard treatment for multiple myeloma patients younger than about 65 years. The role of tandem autologous HSCT for multiple myeloma still remains undefined. Recently, several studies suggested a beneficial role of tandem autologous HSCT for multiple myeloma patients with poor prognosis [7].
PTLD is one of the serious complications following solid organ or HSCT and include a wide range of diseases from benign hyperplasia to malignant lymphomas. PTLD usually develops within the first year after transplantation and more than 80% cases are associated with EBV. In a setting of transplantation, EBV-specific cytotoxic T lymphocytes, which regulate EBV-infected B cell proliferation, are strongly suppressed. This impaired immune surveillance for EBV is assumed to result in the development of PTLD. In addition to EBV infection, other contributing factors, such as allogeneic antigens and cytokines are necessary for the development of PTLD. The incidence of PTLD in allogeneic HSCT has been reported between 0.5% and 2.5% [1], with peak incidence between 2 to 6 months after transplantation. The risk of PTLD after allogeneic HSCT is associated with T-cell depletion of the donor marrow, Antithymocyte Globulin (ATG) use, presence of acute or chronic graft-versus-host disease, unrelated and/or HLA mismatched grafts, age 50-years or older at transplantation [8].
Classical HL type PTLD is the rarest form of PTLD and histopathologically identical to classical HL, showing RS cells in various inflammatory cells. Immunophenotypically, the RS cells express both CD15 and CD30. In a large retrospective analysis of 18,351 patients receiving allogeneic HSCT, a total of eight cases developed HL. Mixed cellularity was the most commonly reported subtype and five of six cases contained EBV genome. These cases differ from typical PTLD on later onset (> 2.5 years after HSCT), generally good prognosis, and the absence of previously reported risk factors, such as T-cell depletion of donor marrow, HLA mismatched and/or unrelated donor, and use of ATG [3].
HL-like PTLD is grouped with classic HL type PTLD because of its morphologic resemblance, including the presence of RS-like cells. Immunohistochemically, RS-like cells express CD20, CD30, but not CD15 [9], whereas RS cells in HL type PTLD express CD15, CD30, and occasionally CD20. Classic HL type PTLD are positive for EBER, limited to the RS cells, but lack EBER in a subset of small-sized background lymphocytes. In contrast, in HL-like PTLD, EBER positivity is observed in both RS-like cells and small background lymphocytes [10]. Clinically, HL-like PTLD can involve nodal or extranodal sites such as lung and/or liver. The time from transplantation to development of PTLD varies from about 1 year to 20 years after transplantation [9,10]. The majority of HL-like PTLD indicate more aggressive behavior [9,11]. These differences between classical HL type PTLD and HL-like PTLD suggest that HL-like PTLD represents a distinct PTLD with overlapping feature to classical HL type PTLD [10,11]. There is no standard approach to the treatment of HL-like PTLD. Because the majority of HL-like PTLD cases exhibited aggressive behavior, current reports suggested that systemic chemotherapy and/or immunotherapy, such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine), should be considered early in the course of those patients [9,10]. In our patient, ABVD cannot be used because of severe liver damage. Mini-BEAM (carmustine, etoposide, cytarabine, melphalan) is often used as salvage therapy for HL and less likely to cause a liver damage. In Japan, carmustine is not available, and ranimustine has been used instead of carmustine (mini-MEAM). Therefore, the patient was treated with mini-MEAM regimen.
Compared to the allogeneic setting, the development of PTLD is very rare complication in autologous HSCT [2], perhaps due to the fact that these patients are less immunosuppressed than patients who received solid organ transplantation or allogeneic HSCT. Furthermore, HL type PTLD occurring after autologous HSCT is particularly rare. To date, only four cases have been reported. All except one cases developed HL 2 to 7 years after the transplantation and contained EBV in the neoplastic cells [4,6]. In consideration of later onset after autologous HSCT and the absence of continuous immune impairment, these cases may not correspond to a real PTLD. On the other hand, Zambelli, et al. reported the patient who developed EBV-negative nodular sclerosis type of HL at day +105 after HSCT for malignant glioma. They described that the rapid onset of HL, the clinical and histopathological characteristics are consistent with a presentation of HL as PTLD [5].
In our case, RS-like cells express CD20, CD30, but not CD15, and EBER positivity is observed in both RS-like cells and background lymphocytes. In contrast, RS cells in HL type PTLD express CD15, CD30, and occasionally CD20 and are positive for EBER, limited to the RS cells. And RS cells in classical HL usually express CD15, CD30, and occasionally CD20. Rowlings, et al. described that post-transplant HL was characterized by late onset (> 2.5 years after bone marrow transplantation) [3]. The patient developed HL-like PTLD at relatively early time after tandem autologous HSCT. Therefore, we speculate that prolonged impairment of cellular immunity after tandem autologous HSCT played a role in emergence of EBV-associated HL-like PTLD through suppression of EBV-specific cytotoxic T lymphocytes.
Hepatic infiltration by HL cells usually develops late in the course of the disease or with advanced stage disease. Several mechanisms have been implicated for acute liver dysfunction in lymphoma. Involvement of small intrahepatic bile ducts results in extensive cholangitis, duct necrosis, and acute liver failure [12]. Infiltration of sinusoids or obstruction of hepatic vasculature by HL cells causes diffuse hepatic necrosis [13]. Cytokines released from HL cells may cause interlobular bile duct destruction and portal fibrosis [14]. The presence of mononuclear variants of RS cells is enough for a diagnosis of hepatic involvement if the diagnosis of HL has been established in a lymph node [15]. The prognosis with liver involvement is usually poor. Early diagnosis and initiation of chemotherapy can reverse the hepatic dysfunction and lead to improve the prognosis.
In conclusion, we report a case of HL-like PTLD that developed following tandem autologous HSCT for multiple myeloma. HL-like PTLD likely represents a separate entity with an aggressive clinical course. Distinction between HL-like PTLD and HL type PTLD is important for clinical management.
References
- Singavi AK, Harrington AM, Fenske TS (2015) Post-transplant lymphoproliferative disorders. Cancer Treat Res 165: 305-327.
- Socie G, Baker KS, Bhatia S (2012) Subsequent malignant neoplasms after hematopoietic cell transplantation. Biol Blood Marrow Transplant 13: 1121-1134.
- Rowlings PA, Curtis RE, Passweg JR, et al. (1999) Increased incidence of Hodgkin's disease after allogeneic bone marrow transplantation. J Clin Oncol 17: 3122-3127.
- Fend F, Martinez A, Quintanilla Martinez L, et al. (2002) Clonally unrelated Hodgkin's disease following autologous stem cell transplant for B-cell lymphoma. Br J Haematol 116: 329-333.
- Zambelli A, Lilleri D, Baldanti F, et al. (2005) Hodgkin's disease as unusual presentation of post-transplant lymphoproliferative disorder after autologous hematopoietic cell transplantation for malignant glioma. BMC Cancer 5: 109-113.
- Kulcsar I, Szanto A, Varoczy L, et al. (2012) Hodgkin's lymphoma developed after autologous stem cell transplantation for multiple myeloma. Pathol Oncol Res 18: 733-736.
- Cavo M, Salwender H, Rosifcrf L, et al. (2013) Double vs. single autologous stem cell transplantation after bortezomib-based induction regimens for multiple myeloma: an integrated analysis of patient-level data from phase European III studies. Blood 122: 767.
- Landgren O, Gilbert ES, Rizzo JD, et al. (2009) Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 113: 4992-5001.
- Krishnamurthy S, Hassan A, Frater JL, et al. (2010) Pathologic and clinical features of hodgkin lymphoma-like posttransplant lymphoproliferative disease. Int J Surg Pathol 18: 278-285.
- Ranganathan S, Webber S, Ahuja S, et al. (2004) Hodgkin-like posttransplant lymphoproliferative disorder in children: does it differ from posttransplant hodgkin lymphoma? Pediatr Dev Pathol 7: 348-360.
- Pitman SD, Huang Q, Zuppan CW, et al. (2006) Hodgkin Lymphoma-Like Posttransplant Lymphoproliferative Disorder (HL-Like PTLD) simulates monomorphic B-Cell PTLD both clinically and pathologically. Am J Surg Pathol 30: 470-476.
- Dich NH, Goodman ZD, Klein MA (1989) Hepatic Involvement in Hodgkin's Disease, Clues to Histologic Diagnosis. Cancer 64: 2121-2126.
- Zafrani ES, Leclercq B, Vernant JP, et al. (1983) Massive blastic infiltration of the liver: a cause of fulminant hepatic failure. Hepatology 3: 428-432.
- Hubscher SG, Lumley MA, Elias E (1993) Vanishing bile duct syndrome: a possible mechanism for intrahepatic cholestasis in Hodgkin's lymphoma. Hepatology 17: 70-77.
- Ortfn X, Rodrfguez-Luacu M, Bosch R, et al. (2010) Acute liver failure as the first manifestation of very late relapsing of Hodgkin's disease. Hematol Rep 2: e5.
Corresponding Author
Masaaki Tsuji, Department of Hematology and Immunology, Ohtsu Red Cross Hospital, 1-1-35, Nagara, Otsu-shi, Siga, Japan, Tel: 077-522-4131, Fax: 077-525-8018.
Copyright
© 2017 Tsuji M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.