Molecular Characterization of Novel Porcine Circovirus 3 (PCV3) in Pig Populations in the North of Vietnam
Abstract
In 2015, a novel circovirus (Porcine circovirus 3, PCV3) was detected for the first time from pigs suffered from porcine dermatitis and nephropathy syndrome and reproductive failure. Since then, PCV3 has been reported in several pig producing countries. This study was carried out in order to investigate the presence and further genetic characterization of PCV3 in the pig populations in northern Vietnam. The screening PCR detected the presence of PCV3 in 6 out of 135 samples (4.44%) which were collected from seven northern provinces in 2011 and 2016-2017. The capsid-coding gene (ORF2, 645 nucleotides in length) was successfully sequenced from 5 out of 6 field strains. Compared to a highly diverse PCV3 strain (GD2016-1, KY421347) 5 Vietnamese PCV3 strains contained 39 point nucleotide mutations and 9 of those were non-synonymous. The Bayesian phylogenetic analysis on the basic of ORF2 revealed that PCV3 evolved at a comparable evolutionary rate of the pathogenic PCV2 (2.284 × 10-3 and 1.440 × 10-3, respectively). Besides, this analysis suggested PCV3 could be separated into PCV3a and PCV3b groups, of which the majority of Vietnamese PCV3 strains belong to PCV3a (sub-cluster a1).
Keywords
Porcine circovirus 3, Genetic analysis, Northern Vietnam
Introduction
Porcine circovirus (PCV) is a member of the genus circovirus, family Circoviridae [1]. In pigs, it is acknowledged that Porcine circovirus type 1 (PCV1) is non-pathogenic while Porcine circovirus type 2 (PCV2) is the etiology of a devastating disease of swine, the porcine circoviral disease (PCVD) [2]. To date, PCVD is emerged in almost all swine producing countries [3], including Vietnam [4]. In 2015, in North Carolina (USA), a novel species of the genus circovirus which named Porcine circovirus 3 (PCV3) was detected for the first time from pigs suffered from porcine dermatitis and nephropathy syndrome and reproductive failure [5]. Because of the novelty, PCV3 is now a topic of interest for researchers in the world. As the result, the virus was demonstrated in many countries, such as: USA [5], China [6], Korea [7], Poland [8], and Italy [9], etc. Up to now, there is not available information on the prevalence of PCV3 in pig populations in Vietnam. Thus, this study aimed to investigate the circulation and molecular characterization of the virus in northern Vietnam.
Material and Methods
Tissue samples
All of pooled organs (lymph nodes, heart, lung, spleen, and kidney) used in this study were collected from pigs having different clinical signs, such as: Wasting, diarrhea, respiratory signs, etc. The samples were either archived (n = 60 in 2011) or current (n = 68 in 2016, n = 7 in 2017). The sampling locations were indicated in Table 1.
PCR- based detection of PCV3
Total DNA was extracted from 10% tissue suspensions. In brief, the samples were: i) Lysed (250 µl) in the solution of sucrose/proteinase K (500 µl) at 56 ℃/90 min; ii) DNA phase separated by phenol-chloroformisoamyl (200 µl); iii) DNA pelleted by addition of isopropyl at -20 ℃/15 min; iv) Washed by ethanol 70%; v) Semi-dried and then dissolved in 30 µl of TE buffer (pH 8.0). Centrifugation (12000 rpm/ 15 min at 4 ℃) was applied between steps (ii) to (iv). This study used i) Hot start PCR kit (i-StarMaster, iNtRON Biotechnology, Korea) and ii) Pathogen- specific primers [6] for detection and sequencing of PCV3. List of primers were given in Supplementary Table 1. The thermal profile of PCR was initial denaturation at 94 ℃/5 min; repeated 35 cycles of 94 ℃/30 sec, 55 ℃/30 sec, 72 ℃/90 sec. PCR products were stained by Red safe DNA nucleic acid staining solution (iNtRON Biotechnology, Korea) and analyzed by agarose electrophoresis.
Sequencing of ORF2 gene
Purified PCR products were submitted to 1st BASE (Singapore) and were sequenced in both directions by Sanger sequencing method. The sequences were then assembled by BioEdit v7.1.3.0 [10]. This study successfully recovered five complete ORF2 genes of PCV3 from pooled organs.
For genetic analysis and Bayesian phylogenetic inference of PCV3, a dataset (n = 86) of complete ORF2 sequences, including those deposited in Gen Bank, was collected. Because Bayesian phylogenetic inference uses molecular clock model to infer a time-scaled tree from heterochronous sequences, ORF2 sequences without sampling years were excluded. The complete list of sequences used in this study was shown in Supplementary Table 2.
Calculating genetic distance
Genetic distance between field strains of PCV3 (n = 86, Supplementary Table 2) were calculated on the basic of ORF2 gene. The p-distance was estimated by MEGA7 program [11]. The genetic distance between strains were then displayed as frequency histogram.
Testing for temporal signal in sequence alignment
This analysis was done by TempEst tool [12] which requires a phylogeny without molecular clock and having sampling dates for all of the leaves. In this study, that tree was inferred under assumption of the p-distance and neighbour-joining method which are implemented in MEGA7 program [11]. The 'Root-To-Tip' analysis panel was then selected to visualize a linear regression of root-to-tip genetic distance against sampling time.
ORF2- based Bayesian phylogenetic analysis
The Bayesian coalescent-based Markov chain Monte Carlo (MCMC) analysis was applied under the assumption of a codon-based SRD06 nucleotide substitution model [13] in combination with i) Model of uncorrelated lognormal relaxed molecular clock and ii) Two demographic coalescent models (constant population size, and extended Bayesian skyline plot). In each analysis, the MCMC chain (100 million generations, sampling every 10000 iterations) was performed using the BEAST package v1.7.4 [14]. The output log files were subsequently analyzed in Tracer v1.6 to assess the convergence (effective sample size > 100) and compare between coalescent models using AICM approach [15]. The generated trees were analyzed by Tree Annotator v1.7.4 to find the maximum clade credibility (MCC) tree. That tree was visualized by Fig Tree v1.4.3 program.
Up to date, there are several publications proposed the classification of PCV3, but using inconsistent terminology [6,16,17]. This study followed a scheme which classified PCV3 based on tree topology and genetic markers at codons 24, 27, 77 and 150 of the ORF2 [17]. Accordingly, PCV3 was divided into groups of PCV3a (sub-clusters a1, a2) and PCV3b (sub-clusters b1, b2).
Results and Discussion
The frequency of PCV3 in northern Vietnam
Though the first clinical case due to PCV3 was described in 2015, throughout metagenomic analysis, circo viruses have been known in different species, including pigs [18]. As the result, this study attempted to detect PCV3 in archived (2011) and current (2016, 2017) samples of pigs. It was the main focus of this study to investigate the presence/absence of PCV3 in the North of Vietnam, thus the associations between clinical symptoms and status of PCV3 positive/negative were not described for each tested sample. The screening PCR for PCV3 (Table 1) revealed that there were 6/135 (4.44%) positive samples, and the virus was detected in only 2 out of 7 sampling provinces. Noteworthy, PCV3 was present in a sample collected in 2011. However, comparing to the most recent publication about PCV3 in the UK, 2011 was not the earliest date for the detection of PCV3 in pigs since the virus was found in samples as far back as 2002 [19]. Taken together, the detection of PCV3 in archived tissue samples in 2002 and 2011 suggested that the virus had emerged in pig populations several years prior to the first clinical case was described [5]. Though the virus was found at a higher positive rate in pigs with severe respiratory disease than in those with mild respiratory disease [20], it was also detected in clinical healthy pigs [21]. Despite of those controversial results, the wide distribution of PCV3 in many swine producing countries suggests for further studies in order to prove the accordance of PCV3's pathogenicity with the Koch's postulates.
ORF2- based genetic characterization of PCV3 in northern Vietnam
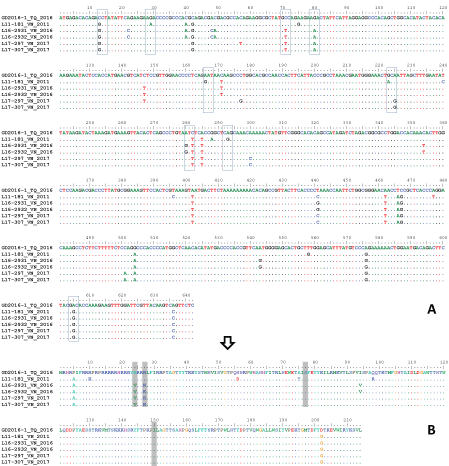
In the PCV3 genome, ORF2 is one of the three predicted open reading frames which encodes protein showing homology to circovirus capsid protein [5]. The nucleotide identities among PCV3 strains were quite similar between ORF2 (96.7%-99.1%) and complete genome (97.5%-99.4%) [22]. Additionally, the topologies of phylogenetic trees based on either complete genome or ORF2 largely agreed with each other [7,22]. That was the reason why ORF2 was selected for molecular characterization of PCV3 in this study. Depicted in Figure 1, the ORF2 of 5 PCV3 strains circulating in Vietnam had similar length of 642 nucleotides (excluding the stop codon). Based on several phylogenetic analyses [20,22], it was observed that GD2016-1 (KY421347) located near the root of a phylogeny. Therefore, at the present, GD2016-1 is believed to exhibit the highest level of nucleotide diversity than other sequences. Comparing to that reference, the ORF2 of 5 PCV3 strains differed in 39 positions, 9 of those were non-synonymous substitutions (boxes, Figure 1A and Figure 1B). There were 4 strains isolated in 2016 and 2017 (L16-2931, L16-2932, L17-297 and L17-307) had a typical genetic marker (V-K-S-I at codons 24, 27, 77 and 150) of sub-cluster a1 [17]. The last isolate (L11-181) had a typical genetic marker of sub-cluster b1 (A-R-S-I at codons 24, 27, 77 and 150) [17].
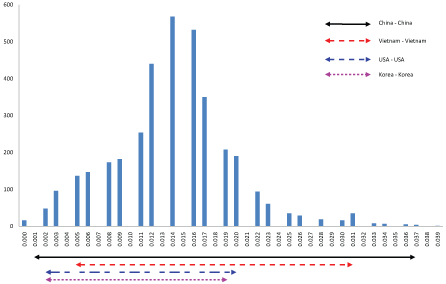
The ORF2 based genetic distance between Vietnamese PCV3 strains and others strain available in Gen Bank were calculated and shown in Figure 2. Excluding 13 identical sequences, the p-distance between 73 unique PCV3 strains were 0.002 to 0.040. In case of porcine circovirus type 2, p-distance of 0.035 was proposed to be the cut-off value for genotype delimitation [23]. Under this point of view, the genetic distance between PCV3 strains was considered large enough. The Chinese PCV3 strains exhibited widest genetic variations (0.002-0.037). The genetic distance between PCV3 strains circulating in each country (Vietnam- Vietnam, USA- USA, and Korea- Korea) was almost within the range of those in China (Figure 2). The result implied that PCV3 might present in the pig populations for a period and might diversify locally.
Evolutionary parameters of PCV3 based on the ORF2 gene
Because of its novelty, details about phylogenetic analyses of PCV3 were not popular. To inferring the genetic relationships between PCV3s, almost all publications to date preferred neighbour-joining method [6,17] and maximum-likelihood method [5,7,20]. However, this study interested in co-estimation of evolutionary rate and the time to the most recent common ancestor (TMRCA), thus, Bayesian coalescent-based MCMC method was applied.
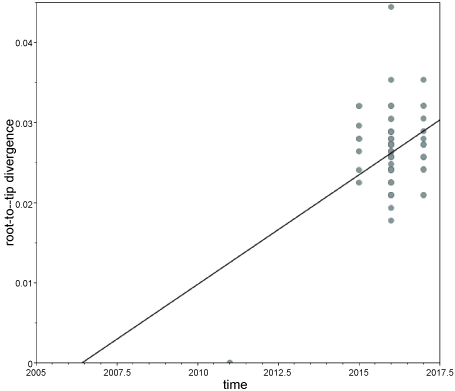
It was due to almost all of PCV3 sequences deposited in Gen Bank were from recent years (2015-2017), the first step in Bayesian phylogenetic analysis is testing whether the dataset contains measurable amounts of evolutionary change between sampling times. Using TempEst tool [12] the ORF2 alignment (listed in Supplementary Table 2) was found to exhibit positive correlation (correlation coefficient, R2 = 0.179) between genetic distances and sampling dates (Supplementary Figure 1). Thus, the current ORF2 dataset appears to be suitable for Bayesian MCMC analysis assuming molecular clock.
Under the uncorrelated log-normal relaxed molecular clock [24], the mean coefficient of correlation of 0.51 suggests that the strict molecular clock could be dismissed. Based on the ORF2 gene, the PCV3 evolutionary rate was estimated at 2.284 × 10-3 substitutions per site per year (95% highest posterior density (HPD) intervals: 1.257 × 10-3 - 3.474 × 10-3). The high evolutionary rate of the capsid coding gene of PCV3 was comparable with that of PCV2 (1.440 × 10-3) [25], a known pathogen which cause porcine circovirus-associated disease. Based on the current dataset which all of PCV3s were sampled from 2011 to 2017, the estimated clock rate gave the TMRCA of PCV3 was approximately 10 years ago (95% HPD intervals: 6.00 - 15.37). However, this result should be interpreted with caution as there was not available sequences of PCV3 from archived samples as far back as 2002 [19]. It is worth to mentioned that the TMRCA reported in this study basically differed from that in a recent publication [16] in which the TMRCA (approximately 50-years-ago) was the divergence time when PCV3 started to separate from a closely related porcine circovirus (PorkNW2/USA/2009, HQ738638).
ORF2- based Bayesian phylogenetic analysis
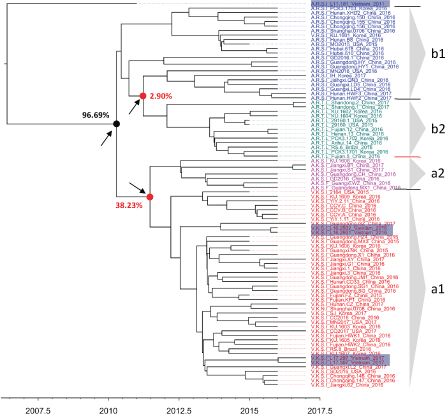
Depicted in Figure 3 is the MCC phylogenetic tree of PCV3 which was inferred on the basic of ORF2 sequence without an out group. It was due to the fact that a relaxed clock model (the uncorrelated lognormal relaxed molecular clock of this study) is able to estimate the position of the root of the tree, even in the absence of a known out group [24]. In agreement with the previous publication [6, 17], the MCC phylogeny supported a separation into distinct groups of PCV3a (51 strains) and PCV3b (34 strains) with highest probability density of 96.69% (solid arrow, Figure 3). However, low posterior probability values were observed at the nodes corresponding to the split into sub-clusters of each PCV3a or PCV3b (dashed arrows, Figure 3). In future studies, uncertainty in genetic relationships between sub-clusters should be refined by different genetic regions and with addition of sequences from archived samples. Of the virus circulating in Vietnam, 4 strains collected in 2016-2017 (highlighted, Figure 3) were grouped within PCV3a (sub-cluster a1), and they all closely related to Chinese PCV3 strains. Although bearing a genetic marker of sub-cluster b1 (A-R-S-I at codons 24, 27, 77 and 150), sample L11-181 (collected in 2011) did not group with either PCV3a or PCV3b. For the phylogenetic relationships of L11-181 with other PCV3 strains to be elucidated, further analyses should be carried out as new sequences are made available.
Conclusions
By confirming the presence of PCV3 in Vietnam, this study added evidence that PCV3 widely distributed in many swine producing countries. On the basic of the ORF2, the Bayesian phylogenetic analysis suggested that PCV3 evolved at a high evolutionary rate of 2.284 × 10-3 substitutions per site per year and the majority of Vietnamese PCV3 strains belong to PCV3a group (sub-cluster a1).
References
- Allan GM, Ellis JA (2000) Porcine circoviruses: A review. J Vet Diagn Invest 12: 3-14.
- Segales J, Allan GM, Domingo M (2005) Porcine circovirus diseases. Anim Health Res Rev 6: 119-142.
- Rose N, Opriessnig T, Grasland B, et al. (2012) Epidemiology and transmission of porcine circovirus type 2 (PCV2). Virus Res 164: 78-89.
- Huynh TM, Nguyen BH, Nguyen VG, et al. (2014) Phylogenetic and phylogeographic analyses of porcine circovirus type 2 among pig farms in Vietnam. Transbound Emerg Dis 61: e25-e34.
- Palinski R, Pineyro P, Shang P, et al. (2017) A novel porcine circovirus distantly related to known circoviruses is associated with porcine dermatitis and nephropathy syndrome and reproductive failure. J Virol 91.
- Ku X, Chen F, Li P, et al. (2017) Identification and genetic characterization of porcine circovirus type 3 in China. Transbound Emerg Dis 64: 703-708.
- Kwon T, Yoo SJ, Park CK, et al. (2017) Prevalence of novel porcine circovirus 3 in Korean pig populations. Vet Microbiol 207: 178-180.
- Stadejek T, Wozniak A, Milek D, et al. (2017) First detection of porcine circovirus type 3 on commercial pig farms in Poland. Transbound Emerg Dis 64: 1350-1353.
- Faccini S, Barbieri I, Gilioli A, et al. (2017) Detection and genetic characterization of Porcine circovirus type 3 in Italy. Transbound Emerg Dis 64: 1661-1664.
- Hall TA (1999) BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Aymposium Series 41: 95-98.
- Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33: 1870-1874.
- Rambaut A, Lam TT, Max Carvalho L, et al. (2016) Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2: 7.
- Shapiro B, Rambaut A, Drummond AJ (2006) Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol 23: 7-9.
- Drummond AJ, Suchard MA, Xie D, et al. (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29: 1969-1973.
- Baele G, Lemey P, Bedford T, et al. (2012) Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol 29: 2157-2167.
- Fu X, Fang B, Ma J, et al. (2017) Insights into the epidemic characteristics and evolutionary history of the novel porcine circovirus type 3 in southern China. Transbound Emerg Dis.
- Fux R, Sockler C, Link EK, et al. (2018) Full genome characterization of porcine circovirus type 3 isolates reveals the existence of two distinct groups of virus strains. Virol J 15: 25.
- Li L, Kapoor A, Slikas B, et al. (2010) Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J Virol 84: 1674-1682.
- Collins PJ, McKillen J, Allan G (2017) Porcine circovirus type 3 in the UK. Vet Rec 181: 599.
- Zhai SL, Zhou X, Zhang H, et al. (2017) Comparative epidemiology of porcine circovirus type 3 in pigs with different clinical presentations. Virol J 14: 222.
- Zheng S, Wu X, Zhang L, et al. (2017) The occurrence of porcine circovirus 3 without clinical infection signs in Shandong province. Transbound Emerg Dis 64: 1337-1341.
- Wen S, Sun W, Li Z, et al. (2018) The detection of porcine circovirus 3 in Guangxi, China. Transbound Emerg Dis 65: 27-31.
- Grau-Roma L, Crisci E, Sibila M, et al. (2008) A proposal on porcine circovirus type 2 (PCV2) genotype definition and their relation with postweaning multisystemic wasting syndrome (PMWS) occurrence. Vet Microbiol 128: 23-35.
- Drummond AJ, Ho SY, Phillips MJ, et al. (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4: e88.
- Firth C, Charleston MA, Duffy S, et al. (2009) Insights into the evolutionary history of an emerging livestock pathogen: Porcine circovirus 2. J Virol 83: 12813-12821.
Corresponding Author
Van Giap Nguyen, D.V.M, MSC, PhD, Department of Veterinary Microbiology and Infectious Diseases, Faculty of Veterinary Medicine, Vietnam National University of Agriculture, Hanoi, Vietnam, Tel: +84-979-008-128.
Copyright
© 2018 Nguyen VG, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.