The Gastric Microbiome of Four Malaysian Gastroduodenal Disease Patients
Abstract
The human gut microbiota has crucial effects on our health and perturbation of the gut microbiome is linked to many human diseases. Within the gut, the highly acidic stomach is an extremely hostile ecological niche for most microbes. The human gastric pathogen, Helicobacter pylori, is the prominent member of the human gastric microbiota. Studies have also demonstrated that other bacteria can also be found in the human stomach. However, the precise composition of the human gastric microbiome is still not fully established. Therefore, this study aimed to investigate the global biodiversity of the human gastric microbiome using the shotgun metagenomics on the gastric biopsies tissues of four Malaysian adults with gastroduodenal disease using the Illumina HiSeq platform. Meta Genome Rapid Annotation using Subsystem Technology (MG-RAST, v3.1) server was used for data analysis. H. pylori was present in all the gastric samples with the lowest abundance in gastric cancer patient. Interestingly, Burkholderia pseudomallei, an "accidental pathogen" that causes melioidosis with high mortality, was also found in the stomach of these patients. The presence of B. pseudomallei in the stomach of asymptomatic human subjects was unknown. In addition, investigation of non-human eukaryota identified a novel Toxoplasma species in surprisingly high abundance in the gastric microbiome. This study highlighted the potential complexity of the human gastric microbiome comprising of various prokaryotes and eukaryotes, as well as different viruses. Findings from this study will serves as a basis for future studies to investigate the interaction between gastric microbiota and human gastroduodenal health and diseases.
Keywords
Human, Gastric microbiome, Helicobacter pylori, Burkholderia pseudomallei, Toxoplasma
List of Abbreviations
GI: Gastrointestinal; GIST: Gastrointestinal Stromal Tumors; HERVs: Human Endogenous Retroviruses; HIV: Human Immunodeficiency Virus; M5NR: M5 Non-Redundant Protein; NGS: Next-Generation Sequencing; UMMC: University of Malaya Medical Centre
Introduction
The human gastrointestinal (GI) tract harbors one of the most complex microbial ecosystems [1,2]. The GI microbiota constitutes mainly of Bacteria and other minority members (Archaea, fungi and viruses). An elucidation of the GI microbiota diversity is vital for the understanding of the roles that microbiota play in host health and for discovering novel approaches to modulate the microbiota for future GI diseases prevention and treatment [3]. The human microbiota is relatively stable, of which disturbances in the GI microbiota have been shown to be associated with diseases [3], such as obesity [4], diabetes [5,6] and inflammatory bowel disease [7], which leads to an increasing interest of the scientific community in this field of research. Hence, the comprehensive genome of these microbial populations possesses far greater genetic potential than the human genome alone [1].
The stomach is a niche environment within the GI tract. Because of the harsh conditions (including low pH), the human stomach was considered sterile until the discovery of the human gastric pathogen, Helicobacter pylori [8]. To date, there is very little information on other microbes of the human gastric microbiota (especially archaea and viruses) and their role in health and diseases [9-15]. Traditionally, the characterization of the human gastric microbiota relied on the cultivation of bacteria from gastric juice or mucosal biopsy [12,13]. However, culture-based methods have considerable limitations, because more often than not, only a small fraction of the microbiota can be cultivated in vitro [16]. The ideal growth conditions and growth requirements for most microorganisms are not well-known, hence only a small proportion of microorganisms in complex communities can be cultivated in the laboratory.
More advance methods evaluate the bacterial diversity based on 16S rRNA gene sequences from a wide variety of microbes obtained directly from gastric samples aided in the identification of bacterial species (including those uncultivable) and revealed the true complexity of the human gastric microbiota [9-11,14,15]. Members from the phyla of Firmicutes, Protobacteria, Actinobacteria and Fusobacteria, as well as yeast at relatively low abundance, have been identified [9,11]. However, 16S rRNA-based methodology is only useful for providing information of bacteria present but cannot tell us about the presence of viruses, protists, and fungi. The rapid development of culture-independent high throughput nucleotide sequencing methods proves us with the opportunity to explore the total genomic composition (metagenome) of the human gastric microbiome in both of taxonomic and metabolic perspectives. Recently, Zhang, et al. (2015) performed whole genome shotgun sequencing directly on endoscopic gastric biopsies from the Weill Cornell Medical College Gastric Cancer and H. pylori Research Database to identify low abundance microbiome of the human stomach [17]. In this pilot study, comprehensive survey of the microbial diversity of human gastric microbiome in four Asian patients with gastroduodenal diseases was carried out using a similar shotgun metagenomic approach.
Materials and Methods
Gastric biopsy samples
Gastric biopsy tissue samples were obtained from antrum and body of the stomach of patients during endoscopic examination at the University of Malaya Medical Centre (UMMC) Endoscopy Unit between August 2011 and January 2012. Tissue samples for histopathological examination were fixed in formalin while those for metagenomic study were stored at -80 °C until DNA extraction. Samples from 4 patients (4 antrum and 4 body tissues) were selected for metagenomic analysis. Simultaneously, blood samples were collected for serological studies.
DNA extraction and DNA sequencing
DNA was extracted from tissue samples using the MasterPure DNA Purification Kit (Epicentre Biotechnologies, Madison, WI, USA) according to manufacturer's instruction. Library was prepared using the Nextera XT Library Prep Kit (Illumina, San Diego, CA, USA). DNA sequencing was performed on the Illumina HiSeq 2500 platform (Illumina). The samples were sequenced on a single flow cell lane as 50-bp paired-end reads.
Data analysis
Although the short read length may mean that a large proportion of unassembled reads might be too short for functional annotation, assembly of the reads might suppress low abundance species. Furthermore, Meta Genome Rapid Annotation using Subsystem Technology (MG-RAST, v3.1) server (http://metagenomics.anl.gov/) is capable of analyzing unassembled Illumina short reads [18]. For initial evaluation of unassembled data, the MG-RAST platform was used. Quality control (QC) filtering for nucleotide sequence data, including removal of artificial duplicate reads, quality-based read trimming and length-based read trimming, was performed on MG-RAST using the default settings prior to annotation [18]. Organism abundance analysis was carried out by similarity search using BLAT against the M5 non-redundant protein (M5nr) database [19]. Translated protein sequences with 60% identity, e-value of < 10-5 with minimum alignment length of 15 amino acids were considered significant matches.
Post-QC reads with length less than 35 bp were removed prior to further analysis. Megan (version 5.10.5) was used to further investigate the diversity of prokaryotes and eukaryotes in the samples [20]. Megan has been demonstrated to be able to assign fragments as short as 35 bp to species [20]. Using Megablast, reads with matches of 90% identity or better and over at least 60 nucleotide match length to the human genome (GRCh38) were taken as human derived. All these reads mapping to the human genome were removed. The remaining non-human reads were mapped using blastn and blastx against the Genbank's peptide (nr) and the nucleic acid datasets (nt) with default blast filters. The output files were loaded into Megan and taxonomic abundances visualized using column charts in Megan. Verification of the most abundant organisms was performed manually. The percentage of genomic non-coding DNA and correlation between blastn and blastx read counts was calculated. Reads mapped to multiple ultraconserved regions of eukaryotes with perfect identity and top hits randomly assigned by Megan were removed from consideration. The unfiltered sequencing reads from this study are available for public access on MG-RAST.
Results and Discussion
Patient background
Among the samples, UM022 was from a patient diagnosed with poorly differentiated adenocarcinoma (linitis plastic) while UM037 was from a patient who suffered from gastrointestinal stromal tumors (GIST) at fundus with no evidence of distal metastasis. The sample, UM067, was derived from a patient antral gastritis. The fourth sample, UM122, was derived from a patient with gastric ulcers and duodenitis. The mean age of the four patients was 67-years-old (64-70 years old) at the time of sampling (August 2011-January 2012). The ratio of male to female subjects was 1:1.
Overview of the human gastric metagenomes
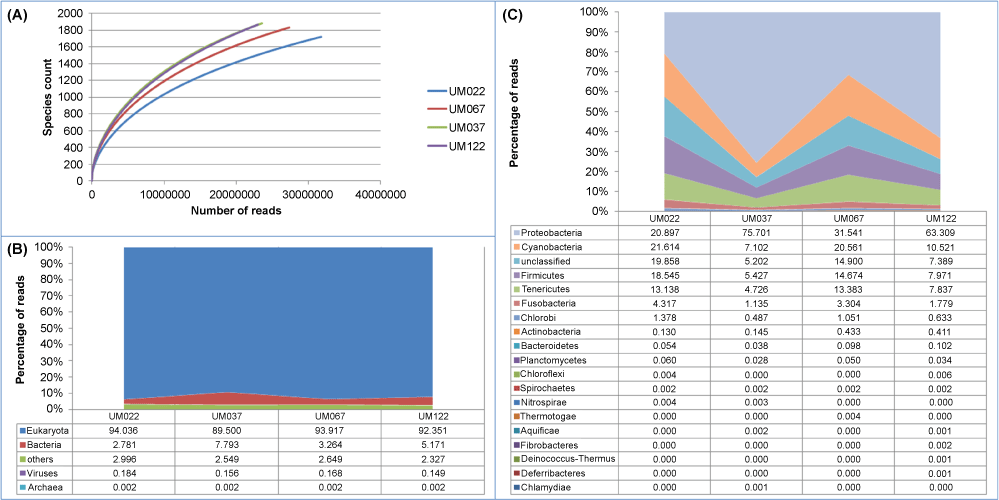
MG-RAST applied a QC filter to remove the lower quality and duplicate reads. Filtering removed about 2.2% of the reads on average. Further analysis used only sequences that passed the initial QC step. The mean number of sequences per sample that passed QC was 20,670,149 reads. The mean read length was 96 ± 3 bp. Table 1 summarized the QC statistics of the metagenomes based on MG-RAST annotation. Figure 1A presents the rarefaction curves of the metagenomes generated by MG-RAST after QC filtering.
Prior to filtering out human sequences, the ensuing identification and abundance indicate that Eukayota (inclusive of both human and non-human sequences) was the most abundant domain (92.4%, mean), followed by Bacteria (4.8%) (Figure 1B). Viruses only accounted for < 0.2% while Archaea constituted < 0.01% of the human gastric microbiome. It is not surprising that Eukaryota is the most abundant as human DNA is expected to predominate in the gastric mucosal biopsies. Unlike samples from mucosal surfaces (such as oral cavities, skin and urogenital tract), which are highly enriched for bacteria, gastric mucosal biopsies obtained through endoscopy by pinching contain mostly human DNA and relatively little bacterial DNA [16]. In addition, food ingested may also contribute to non-human Eurkayota DNA.
To elucidate the bacterial communities, analysis by MG-RAST using the M5NR database classified the human gastric bacterial microbiome into 18 different phyla (Figure 1C). Consistent with many previous reports [9-11,14], the most common phyla of the human gastric microbiome are Proteobacteria (47.9%, mean), Cyanobacteria (14.9%), Firmicutes (11.7%), Tenericutes (9.8%) and Fusobacteria (2.6%). It is interesting to note that while Proteobacteria was the predominant phylum in patients with gastric ulcers/duodenitis and GIST, the proportion of Proteobacteria to other major phyla was more evenly distributed in patients with gastric cancer and gastritis. However, the sample size in the current study is too small to make any meaningful deduction about the link between specific gastric microbiota differences and gastroduodenal diseases.
To characterize the microbial communities of the human gastric microbiome, reads mapping to the human genome were filtered out. In order to better estimate the eukaryote and prokaryote populations, number of reads expressed in reads per million was normalizing for the different genomic length of each species (Table 2 and Table 3). The most abundant organisms found in the human gastric microbiome are shown in (Table 2 and Table 3). Many of these organisms found are also known to be part of the oral and upper GI microbiota. Similarly, the presence oral and upper GI bacteria have also been reported in the stomach of healthy subjects [12]. In addition, since these samples were obtained during gastroendoscopy, we cannot rule out the possibility of contamination by oral and upper GI microbiota during the procedure.
Bacteria
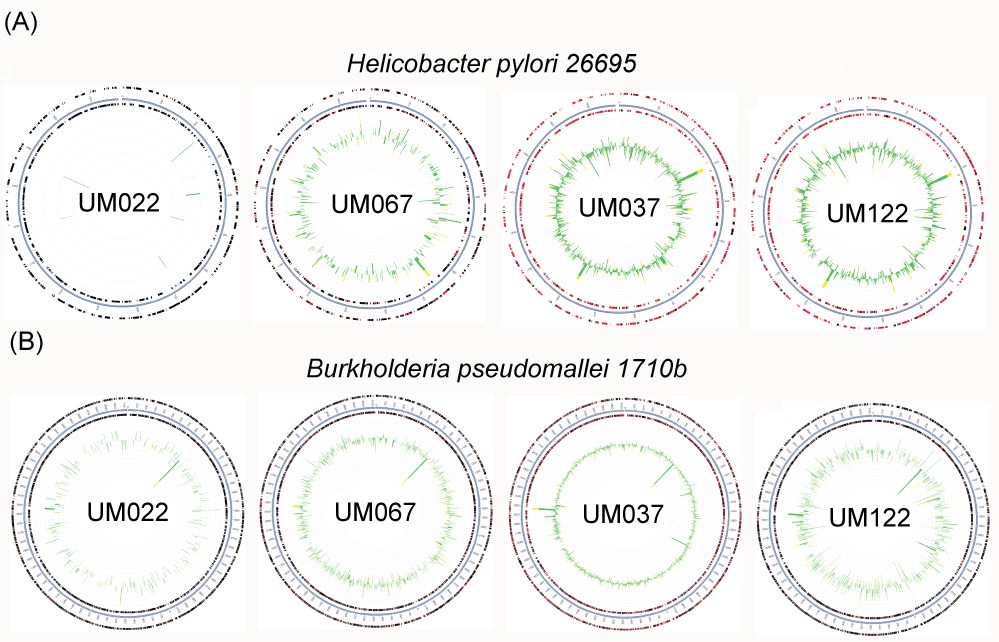
The human gastric pathogen, H. pylori, was lowest in abundance in UM022, a patient with gastric adenocarcinoma (Table 2). This observation was consistent with results from H. pylori culturing. UM022 was negative for H. pylori culture whilst H. pylori was isolated from the other three samples. The metagenomic approach, on the other hand, enhances the detection of this human gastric pathogen in UM022. Indeed, genes that matched those present in the genome of H. pylori were identified. Genes mapped against the standard H. pylori 26695 strain by MG-RAST (Figure 2), provided evidence for the presence of H. pylori in UM022. Such low abundance of H. pylori may indicate that H. pylori almost completely disappeared from the stomach of advanced stage gastric cancer patients probably because of the altered gastric environment in gastric cancer. pH, as well as bacterial count of the gastric environment, have been reported to be elevated in patients with severe atrophic gastritis and gastric cancer [21]. Alternatively, H. pylori may have converted to a viable but non-culturable coccoid form and remains low abundance in UM022 because of the altered gastric environment.
Another interesting observation from our data was the detection of Burkholderia pseudomallei in the stomach of all these Malaysian patients. B. pseudomallei, a soil bacterium, is the etiological agent of melioidosis, an endemic infectious disease in Southeast Asia, northern Australia, Indonesia, Indian subcontinent, southern China, Hong Kong and Taiwan [22]. The recruitment plots (Figure 2) show that reads predicted to be from B. pseudomallei were mapped evenly throughout the B. pseudomallei 1710b genome by MG-RAST. This provides evidence supporting the identification of B. pseudomallei in these samples. In our earlier study, B. pseudomallei was also isolated in gastric biopsy tissues of other Malaysian patients undergoing endoscopy [13]. Coincidentally, Goodyear, et al. had demonstrated in mice that B. pseudomallei preferentially colonize the stomach and the colonized stomach might serve as a reservoir for dissemination of infection to extra-intestinal sites [23]. Tracing their medical history and using an in-house developed test [24], none of the Malaysian patients has melioidosis and they were tested negative for sero-conversion against B. pseudomallei. Consistently, a B. pseudomallei-specific in situ hybridization test [25] to detect for the bacterium in gastric biopsy tissues collected for histopathological examination did not show sign of intracellular invasion by B. pseudomallei. Despite causing melioidosis, we postulate that the bacterium might be part of the gastric microbiome of people from places endemic for B. pseudomallei. The significance of finding the organism in the stomach of human is still unknown but all these evidences suggest that the human stomach might be a niche for persistent harboring of this bacterium without triggering the host immune system.
Viruses
As infective viruses (DNA and RNA viruses) are expected to be integrated into the human genome to utilize the human transcriptional/translational mechanisms for proliferation, filtering out of human sequences from the dataset may result in substantial loss of information regarding viral members of the human microbiome. Thus, analysis for viruses was carried out using data without filtering out for human sequences. Viruses were found to be a minority member of the human gastric microbiome compared to bacteria. However, matching against the M5NR database revealed human viruses, proviruses and bacteriophages in the human gastric microbiome (Table 3). The dominant viral member of the gastric microbiome was Retroviridae, mostly classified as human endogenous retroviruses (HERVs). HERVs are integrated within the human genome and these retrovirus-like sequences have been estimated to make up as much as 8% of our genome [26]. While the ancient retroviruses that overcame our host defense mechanisms and permanently integrated into our genome had been thought to be mostly inactive [27], recent evidence suggest that these viral sequences might be active and have roles in neurological diseases and cancers [28-30]. While it is possible that some of the HERVs are active in the gastric environment, we cannot distinguish the active HERVs from those inactive ones with the current data.
The second most abundant group of virus detected was Herpesviridae. Recently, it was reported that inflammatory bowel diseases patients with Herpesviridae sequences in their colon also demonstrated differences in abundance of human endogenous viral sequences and diversity of their microbiome suggesting interplay between viruses and bacteria in the human gut [31]. A member of the Herpesviridae, Epstein-Barr virus (EBV), has previously been demonstrated to precede malignant transformation in a significant fraction of gastric carcinomas independent of bcl-2 expression and p53 accumulation [32]. EBV was also found in the human gastric mucosal by Zhang, et al. (2015) [16].
Bacteriophages are obligate parasitic agents of specific bacteria and bacteriophages indicate the presence of viable specific bacteria in the environment [33]. The detection of Staphylococcus, Burkholderia and Enterobacteria phages (Table 3) in the human gastric microbiome is consistent with the detection of Staphylococcus aureus, Burkholderia pseudomallei and members of Enterobacteriaceae respectively (Table 2). It remains to be determined whether there is a correlation between the presence of these bacteriophages and clinical presentation of these gastroduodenal diseases or the gastric environment. Bacteriophages can modulate food digestion by regulating microbial communities in the human GI tract through lytic and lysogenic replication [34].
Eukaryota
GenBank NR eukaryote genomes are commonly contaminated with bacteria, and thus blastn may miscall reads as eukaryote when they are bacterial. Using blastx instead of blastn matches abrogates this problem. However, using Megan with blastx to identify eukaryote reads must still be carried out with caution as large percentage of eukaryote genomes contain non-coding DNA sequences, thus the relative abundance of eukaryotes identified by blastx must be corrected by taking into account the percentage of coding sequence in the genome. Another complication of analysis of eukaryotes from metagenome sequencing is many eukaryote genomes were sequenced, but the genome annotation is not sufficiently advanced for the peptides to be deposited in GenBank.
Apart from a large percentage of human reads, we identified other eukaryote reads including fungal (Malassezia sp.), apicomplexan (Toxoplasma sp.) and nematode sequences in all four gastric biopsies using Megan blastx (Table 2). A Toxoplasma species was found in the blastx data with around 80% nucleotide identity to T. gondii, an obligate intracellular, parasitic protozoan (Table 2). This may be a toxoplasma or another closely related unknown apicomplexan. In three of the samples, Toxoplasma was in the top 5 organisms identified in the human gastric microbiome, and as half of the Toxoplasma genome is coding, correcting for this means the apicomplexan was actually the second most abundant in the 4 samples after Staphylococcus. Although gastric toxoplasmosis has rarely been reported and only in cases of severe immunosupression [35-37], findings of T. gondii DNA in all four gastric tissues without human immunodeficiency virus (HIV) suggest that Toxoplasma colonization of the human stomach might not be so rare after all, especially in endemic areas.
Malassezia is a yeast known to be part of the normal human skin microbiota, and, in this case, likely represents yeast shed from the oral cavity [38] into the stomach or an environmental contaminant. It was found in all four gastric biopsy tissue samples.
Our understanding of the prevalence and distribution of microbial eukaryotes in the human gut has significant consequences on human health. This is especially true for populations in developing nations where microbial parasites are large sources of morbidity and mortality [39].
Shotgun metagenomics
Despite that next-generation sequencing (NGS) technologies have become more accessible and economical, shotgun metagenomics like these used in this study remain costly. Thus, the numbers of samples used tend to be small. On the other hand, 16S rRNA-based metagenomics is a more economical alternative but only provides information of bacteria that are present in particular anatomical sites. Although PCR-based 16S rRNA metagenomics has a higher sensitivity, PCR also adds biases [40]. Importantly, the presence of DNA associated with an organism or encoding for a function does not indicate that the viable organism is indeed present or the gene is actually being transcribed. Furthermore, metagenomic study cannot distinguish bacteria residing at the site of study from bacterial cell remnants or those of transient presence. This is especially true for metagenomics of the human gastrointestinal tract that constantly received influx of food, which are non-sterile. In addition, the subjects of the present study are diseased subjects. The lack of healthy subjects as control is a major limitation in experimental design. However, it is difficult to justify subjecting a healthy individual to invasive endoscopic procedure from the ethical viewpoint. Nevertheless, our study has provided a comprehensive coverage of the possible global biodiversity of the human stomach.
Conclusions
The set of dominant gastric bacteria was similar to those found in other populations, but we identified some unique community structures that are indicative of geographical variations. Biodiversity of the human gastric microbiome may be more complex than what was previously known. This study of the human gastric microbiome suggests that the human stomach may serve as niche ecology for long term chronic persistence of pathogens and opportunistic pathogens. In addition to bacterial and archaeal DNA, DNA of human viruses, bacteriophages, human parasites and nematodes were also part of the human gastric metagenome. Although, the sample size of this study is small and the results should be treated with caution, this information deepens our understanding of the human gastric microbial ecology and serves as a reference point for future epidemiological studies and translational applications. These preliminary data also support the need for larger-scale studies on the human gastric microbiome to investigate geographical variations and disease associations.
Declarations
Acknowledgements
This study was funded by the Stanford University-Malaysia Genome Institute-University of Malaya collaboration Grant (55-02-03-1002) and the University of Malaya-Ministry of Education (UM-MoE) High Impact Research (HIR) Grant UM.C/625/1/HIR/MoE/CHAN/13/5 (Account No. H-50001-00-A000033). The funding body has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Ethics approval and consent to participate
All experimental procedures were approved and carried out in accordance to the regulations and guidelines of the UMMC Medical Ethics Committee (Ref. No. 943.2 and 1023.3). Informed written consent was obtained from patients.
Availability of data and material
The unfiltered sequenced reads were deposited as project "Shotgun metagenomics of the human stomach" (ID 5712) with MG-RAST. Metagenome accession numbers are mgm4543593.3 (UM122), mgm4543594.3 (UM022), mgm4543595.3 (UM037) and mgm4543596.3 (UM067).
References
- Maccaferri S, Biagi E, Brigidi P (2011) Metagenomics: key to human gut microbiota. Dig Dis 29: 525-530.
- Okuda S, Tsuchiya Y, Kiriyama C, et al. (2012) Virtual metagenome reconstruction from 16S rRNA gene sequences. Nat Commun 3: 1203.
- Tyakht AV, Kostryukova ES, Popenko AS, et al. (2013) Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun 4: 2469
- Turnbaugh PJ, Ley RE, Mahowald MA, et al. (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027-1031.
- Wen L, Ley RE, Volchkov PY, et al. (2008) Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455: 1109-1113.
- Vaarala O, Atkinson MA, Neu J (2008) The "perfect storm" for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes 57: 2555-2562.
- Ott S, Musfeldt M, Wenderoth DF, et al. (2004) Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 53: 685-693.
- Warren JR, Marshall B (1983) Unidentified curve bacilli on gastric epithelium in active chronic gastritis. Lancet 321: 1273-1275.
- Bik EM, Eckburg PB, Gill SR, et al. (2006) Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA 103: 732-737.
- Dicksved J, Lindberg M, Rosenquist M, et al. (2009) Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J Med Microbiol 58: 509-516.
- Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, et al. (2011) Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J 5: 574-579.
- Delgado S, Cabrera-Rubio R, Mira A, et al. (2013) Microbiological survey of the human gastric ecosystem using culturing and pyrosequencing methods. Microb Ecol 65: 763-772.
- Khosravi Y, Dieye Y, Poh BH, et al. (2014) Culturable bacterial microbiota of the stomach of Helicobacter pylori positive and negative gastric disease patients. Scientific World Journal 2014: 10
- Sung J, Kim N, Kim J, et al. (2016) Comparison of Gastric Microbiota Between Gastric Juice and Mucosa by Next Generation Sequencing Method. J Cancer Prev 21: 60-65.
- Yang I, Woltemate S, Piazuelo MB, et al. (2016) Different gastric microbiota compositions in two human populations with high and low gastric cancer risk in Colombia. Sci Rep 6: 18594.
- Thomas T, Gilbert J, Meyer F (2012) Metagenomics - a guide from sampling to data analysis. Microb Inform Exp 2: 3.
- Zhang C, Cleveland K, Schnoll-Sussman F, et al. (2015) Identification of low abundance microbiome in clinical samples using whole genome sequencing. Genome Biol 16: 265.
- Meyer F, Paarmann D, D'Souza M, et al. (2008) The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9: 386.
- Wilke A, Harrison T, Wilkening J, et al. (2012) The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics 13: 141.
- Huson DH, Auch AF, Qi J, et al. (2007) MEGAN analysis of metagenomic data. Genome Res 17: 377-386.
- Engstrand L, Lindberg M (2013) Helicobacter pylori and the gastric microbiota. Best Pract Res Clin Gastroenterol 27: 39-45.
- Currie BJ, Dance DA, Cheng AC (2008) The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans R Soc Trop Med Hyg 102: S1-S4.
- Goodyear A, Bielefeldt-Ohmann H, Schweizer H, et al. (2012) Persistent gastric colonization with Burkholderia pseudomallei and dissemination from the gastrointestinal tract following mucosal inoculation of mice. PLoS One 7: e37324.
- Chenthamarakshan V, Kumutha MV, Vadivelu J, et al. (2001) Distribution of immunoglobulin classes and IgG subclasses against a culture filtrate antigen of Burkholderia pseudomallei in melioidosis patients. J Med Microbiol 50: 55-61.
- Eu LC, Ong KC, Hiu J, et al. (2014) In situ hybridization to detect and identify Burkholderia pseudomallei in human melioidosis. Mod Pathol 27: 657-664.
- Belshaw R, Pereira V, Katzourakis A, et al. (2004) Long-term reinfection of the human genome by endogenous retroviruses. Proc Natl Acad Sci USA 101: 4894-4899.
- Brady T, Lee YN, Ronen K, et al. (2009) Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev 23: 633-642.
- Solyom Szilvia, Kazazian H Haig (2012) Mobile elements in the human genome: implications for disease. Genome Med 4: 12.
- Wildschutte JH, Williams ZH, Montesion M, et al. (2016) Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc Natl Acad Sci USA 113: E2326-E2334.
- Pasquarella A, Ebert A, Pereira de Almeida G, et al. (2016) Retrotransposon derepression leads to activation of the unfolded protein response and apoptosis in pro-B cells. Development 143: 1788-1799.
- Wang W, Jovel J, Halloran B, et al. (2015) Metagenomic analysis of microbiome in colon tissue from subjects with inflammatory bowel diseases reveals interplay of viruses and bacteria. Inflamm Bowel Dis 21: 1419-1427.
- Gulley ML, Pulitzer DR, Eagan PA, et al. (1996) Epstein-Barr virus infection is an early event in gastric carcinogenesis and is independent of bcl-2 expression and p53 accumulation. Hum Pathol 27: 20-27.
- Weinbauer MG (2004) Ecology of prokaryotic viruses. FEMS Microbiol Rev 28: 127-181.
- Gorski A, Weber-Dabrowska B (2005) The potential role of endogenous bacteriophages in controlling invading pathogens. Cell Mol Life Sci 62: 511-519.
- Alpert L, Miller M, Alpert E, et al. (1996) Gastric toxoplasmosis in acquired immunodeficiency syndrome: antemortem diagnosis with histopathologic characterization. Gastroenterology 110: 258-264.
- Ganji M, Tan A, Maitar MI, et al. (2003) Gastric toxoplasmosis in a patient with acquired immunodeficiency syndrome. A case report and review of the literature. Arch Pathol Lab Med 127: 732-734.
- Merzianu M, Gorelick SM, Paje V, et al. (2005) Gastric toxoplasmosis as the presentation of acquired immunodeficiency syndrome. Arch Pathol Lab Med 129: e87-e90.
- Dupuy AK, David MS, Li L, et al. (2014) Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: discovery of Malassezia as a prominent commensal. PLoS One 9: e90899.
- Kaplan JE, Jones JL, Dykewicz CA (2000) Protists as opportunistic pathogens: public health impact in the 1990s and beyond. J Eukaryot Microbiol 47: 15-20.
- Brooks JP, Edwards DJ, Harwich MD Jr, et al. (2015) The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol 15: 66.
Corresponding Author
Jamuna Vadivelu, Department of Medical Microbiology, University of Malaya, Malaysia.
Copyright
© 2017 Khosravi Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.