Pseudohypoparathyroidism-Literature Update 2018
Abstract
Pseudohypoparathyroidism (PHP) is a rare heterogenous group of disorders characterized by end-organ resistance to PTH, specifically at the proximal renal tubule. The term PHP encompasses PHP 1A, PHP 1B, PHP 1C, PHP 2 and pseudopseudohypoparathyroidism (PPHP). The two most common subtypes are PHP 1A and PHP 1B, PHP 1A, PHP 1C and PPHP have the Albrights hereditary osteodystrophy (AHO) phenotype, in addition to end-organ resistance to PTH in the two former subtypes. Recently, the European Pseudohypoparathyroidism (EuroPHP) network have proposed a novel classification of PHP which encompasses all disorders of the PTH receptor and its signaling pathway, known as inactivating PTH/PTH-related protein signaling disorders (iPPSD) [1-6].
PHP is caused by molecular alterations within the GNAS locus. The GNAS gene encodes an alpha subunit of the stimulatory G protein (Gsα) that mediates the actions of PTH and other hormones including TSH and gonadotropins. It is a defect in these stimulatory G-protein coupled receptors, that results in end-organ resistance to parathyroid hormone and select other hormones. Mutations of the GNAS gene are associated with iPPSD2 (PHP 1A, PHP 1C, PPHP, progressive osseous heteroplasia (POH)) whereas methylation defects are identified at the GNAS locus and associated with iPPSD3 (PHP 1B, including autosomal dominant-PHP 1B and sporadic-PHP 1B).
This year, the first international consensus statement on the diagnosis and management of pseudohypoparathyroidism was published and is informative to physicians with limited experience treating these rare disorders. It is important to establish a diagnosis to enable appropriate medical treatment of the endocrine complications and provide accurate genetic and prenatal counselling of individuals and their first-degree relatives.
Keywords
Hormone Resistance, Pseudohypoparathyroidism, Pseudopseudohypoparathyroidism, Albrights Hereditary Osteodystrophy, GNAS gene, Chromosome 20q13.3, iPPSD (Inactivating PTH/PTH-Related Protein Signaling Disorders)
Introduction
Pseudohypoparathyroidism (PHP) was first described by Dr. Fuller Albright in 1942 [1]. PHP is an orphan genetic disorder characterized by hypocalcemia, hyperphosphatemia, normal vitamin D 25OH, low vitamin D 1,25OH levels and an elevated PTH level, related to PTH resistance at target organs [2]. The exact prevalence of PHP is unknown [3]. The estimated prevalence in Denmark is 1.1/100,000 [4] and in Japan is 0.34/100,000 [5].
The term PHP encompasses PHP 1A, PHP 1B, PHP 1C, PHP 2 and pseudopseudohypoparathyroidism (PPHP). The original classification of PHP assigned sub-types based on the presence or absence of Albrights hereditary osteodystrophy (AHO), PTH resistance, and the degree of alpha subunit of the stimulatory G protein (Gsα) activity using in vitro assays [6,7] (Table 1). An updated classification of PHP disorders has been proposed by European Pseudohypoparathyroidism (EuroPHP) network that classifies patients based on their clinical diagnosis [8]. The diagnosis of PHP and related disorders is always initially a clinical diagnosis, which can be followed by molecular genetic studies, which confirm the diagnosis and allow characterization of the subtype [9].
Patients with PHP present with symptoms and signs of resistance to PTH, occasionally TSH (PHP 1A, PHP1c and some PHP 1B), follicle stimulating hormone & luteinizing hormone (PHP 1A, PHP 1C) and growth hormone releasing hormone (PHP 1A) [10]. Additionally, AHO is present in patients with PHP 1A, PHP 1C and PPHP but is absent in patients with PHP 2. A mild form of the AHO phenotype can be seen in PHP 1B [3]. PHP is typically diagnosed in infancy or adolescence but there are case reports of PHP being diagnosed in adulthood, particularly PHP 1B and PPHP [11,12].
The genetic defects underlying PHP occurs at the GNAS locus on chromosome 20q13.2-q13.3, where the genes encoding Gsα, XLαs and NESP55 lie, as well as two non-translated transcripts called A/B and GNAS-AS1 (GNAS complex locus, Antisense Transcript 1) [13]. PTH, along with other select hormones, act through these G proteins coupled receptors whose signal transduction pathways is mediated by the alpha subunit of the stimulatory G protein (Gsα).
Most clinicians have limited experience or expertise in treating these specialized disorders. Fortunately, the first international consensus statement on the diagnosis and management of PHP and related disorders was recently published and will help guide clinicians in treating this orphan disorder [3].
This paper will focus on the pathophysiology, differential diagnosis, genetic diagnosis, evolving classification, clinical features and management of PHP in 2018.
PTH Pathophysiology
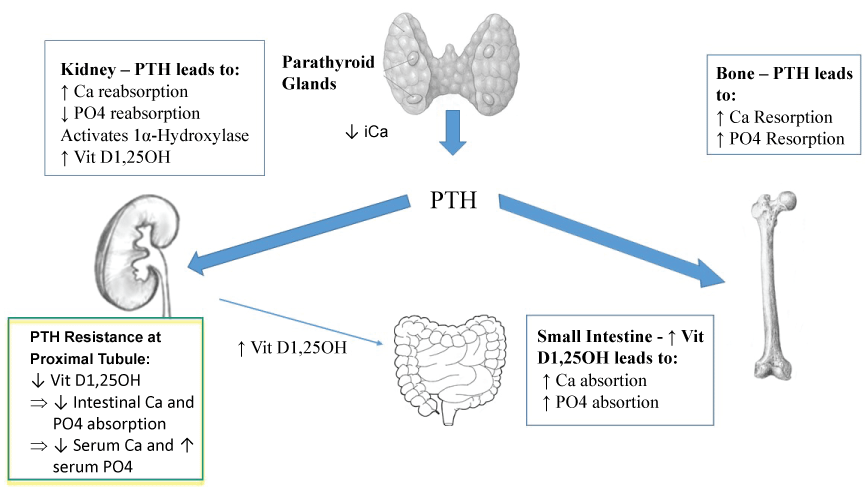
PTH is the predominant hormonal regulator of calcium in the human body and acts primarily at the level of the kidney, bone and gastrointestinal tract (Figure 1). PTH is released from the parathyroid glands in the setting of low ionized calcium levels. PTH mobilisers calcium from bone, increases intestinal calcium absorption by activation of 1-α-hydroxylase in the kidney with increased production of activated Vit D 1,25OH and promotes calcium reabsorption in the distal renal tubule. Additionally, PTH prevents phosphate reabsorption in the renal tubule, thereby maintaining a normal serum phosphate level [10].
The hallmark pathophysiologic defect in PHP is renal PTH resistance; skeletal responsiveness to PTH remains intact [10]. The defect is located in the proximal renal tubule where 1-α-hydroxylase converts Vit D 25OH to Vit D 1,25OH. Low Vit D 1,25OH levels lead to hypocalcemia, as Vit D 1,25OH is necessary for intestinal calcium absorption [10]. Additionally, reduced urine phosphate excretion leads to hyperphosphatemia [10]. Fortunately, the distal renal tubule remains responsive to PTH and urine calcium reabsorption is preserved [14], thereby reducing the risk for hypercalciuria or renal stone formation, which are features of classic hypoparathyroidism. Skeletal responsiveness to PTH is not affected, therefore high PTH levels for prolonged periods of time increases the risk for hyperparathyroid-related bone disease, including osteitis fibrosa cystica [15,16].
The PTH receptor (PTHR1) is coupled to the Gsα protein signal transduction pathway. Normally PTH-mediated activation of this pathway results in an increased production of serum and urine cyclic adenosine monophosphate (cAMP) [17]. Renal tissue resistance to PTH at PTHR1 in the proximal tubule manifests as a reduced urine phosphate and cAMP excretion [17], whereas resistance to PTHrP at the bone could be responsible for the skeletal abnormalities associated with PHP, both due to the haploinsufficiency of Gsα coding GNAS gene [3].
Differential Diagnosis
PHP is characterized by hypocalcemia and hyperphosphatemia with an elevated PTH level, in the absence of vitamin D deficiency or impaired renal function. The differential diagnosis includes vitamin D deficiency or resistance, renal failure, hypomagnesemia, acute pancreatitis, rhabdomyolysis, tumor lysis, osteoblastic metastasis and hyperventilation [18]. The most common etiology that mimics PHP, with low calcium and high PTH levels, is vitamin D deficiency [19,20]. Hypomagnesemia is a recognized reversible cause of functional hypoparathyroidism and is increasingly seen with the widespread use of proton pump inhibitors [21]. The differential of PHP according to phenotype is broad and includes many endocrine and syndromic disorders, which have been described in detail in the recent international consensus paper on PHP [3].
Genetics
A genetic cause can be identified in an estimated 80-90% of patients with PHP, including both sporadic or autosomal dominant heterozygous inactivating mutations within the GNAS gene or epigenetic alterations at the GNAS locus [9], located on chromosome 20q13.2-q13.3 [3]. The imprinted GNAS gene consists of 13 exons that encode the signaling protein Gsα, a ubiquitous signaling protein mediating the actions of many hormones via generation of the second messenger cAMP. Even if Gsα is biallelically expressed in the majority of tissues, maternal Gsα expression is silenced in tissues with maternal imprinting [22] namely the proximal renal tubules, thyroid, gonads and pituitary somatotrophs [13,23]. This tissue-specific imprinting leads to a parent of origin mutation effect: Maternal transmission of the mutations involves the development of AHO and multi-hormone resistance (namely, PHP1A and 1C), whereas paternal transmission is exclusively associated to the phenotypic features of AHO, without any hormone resistance (namely, PPHP and progressive osseous heteroplasia (POH)) [3,24].
PHP1B is caused by loss of imprinting at GNAS locus, affecting either GNAS A/B: TSS-DMR exclusively or in combination with any of the other three differentially methylated regions (DMRs) (GNAS-NESP: TSS-DMR, GNAS-AS1: TSS-DMR or GNAS-XL: Ex1-DMR) [3,9]. In most cases of PHP1B these methylation alterations occur sporadically without any underlying genetic defect, but some cases are related to heterozygous deletions within STX16 or NESP/AS that imply the loss of methylation at GNAS A/B: TSS-DMR alone or at the four DMRs [3,13,23]. Genetic defects at GNAS, observed in patients with PHP 1A are associated with a generalized deficiency or reduction in activity of Gsα [7,25], whereas a slightly diminished activity has been observed in patients with PHP 1B [26].
Today, the majority of these patients with a clinical diagnosis of PHP are candidate for molecular genetic testing. Molecular diagnosis of suspected PHP cases must include DNA sequence, methylation and copy number variant analyses at the GNAS locus [3]. Molecular diagnosis of suspected PHP cases must include DNA sequence, methylation and copy number variant analyses at the GNAS locus [3].
Classification of PHP
The clinical and molecular overlap among the spectrum of PHP and related disorders represents a challenge for the clinician. The current classification of PHP is based on the presence or absence of the AHO phenotype, specific biochemical profiles, nephrogenic cAMP response to exogenous PTH and the in vitro assay measurements of Gsα protein activity [8]. The current classification fails to include related disorders of the PTH/PTHrp signaling pathways such as acrodysostosis, POH and PTH1R-related chondrodysplasia [8]. The EuroPHP network have proposed an updated classification for PHP, that encompasses all disorders with impairments in the PTH and/or PTHrp cAMP mediated pathways, thereby eliminating the clinical or molecular overlap between diseases. This novel classification is called inactivating PTH/PTH-related protein signaling disorders (iPPSD) 1-6 [8], (Table 2). This new classification stratifies the disorders into clusters caused by the same mechanism, thereby simplifying the concept of the overlapping disorders.
Clinical Features
The presentation and severity of PHP vary greatly between affected individuals, with considerable clinical and molecular overlap between the different PHP subtypes. Large inter-familial and intra-familial variability has been observed for the same mutation. There is no correlation between the type or location of the GNAS mutations and the disease onset, degree of hormone resistance or AHO phenotype [3]. In patients with PHP, resistance to hormones namely PTH, TSH, gonadotropins and GHRH are variably present and attributable to genetic imprinting [27,28]. The typical presentation of pseudohypoparathyroidism by age, in the pediatric population, is presented in Table 3. The first international consensus panel on PHP recommend that the diagnosis of PHP be based on major criteria, namely resistance to PTH, ectopic ossification, brachydactyly/AHO and early onset (< 2 years) obesity +/- TSH resistance [3]. The diagnosis of PHP is primarily a clinical diagnosis which should be followed up with genetic testing to confirm the clinical diagnosis and allow characterisation of the particular sub-type.
AHO is a term that describes a number of skeletal and developmental abnormalities seen in PHP. These include shortened metacarpals & metatarsals (brachydactyly), ectopic soft tissue or dermal ossifications, short stature, round facies and cognitive impairment [10]. The short stature is attributable to premature closure of growth plates and short bones [3]. Brachydactyly type E, which is shortening of III, IV and V metacarpals or metatarsals and first distal phalanx (wider than long, also known as potters' thumb), is a classic finding with the AHO phenotype [29] however may not be evident in early life and may develop overtime [3]. Interestingly, the AHO obesity phenotype occurs primarily in those individuals who have multiple hormone resistances and only when the mutation is on the maternal allele, namely PHP 1A [30].
Patients with PHP typically present with the classic symptoms of hypocalcemia, related to PTH resistance; which is seen in 45-80% of cases [3]. This hypocalcemia related to PTH resistance occurs in the absence of vitamin D deficiency. The symptoms of hypocalcemia can range from the asymptomatic, to mild numbness and paresthesias, to more severe life-threatening signs, with the development of laryngospasm, tetany and fatal cardiac arrhythmias [31]. The two hallmark physical exam findings seen with hypocalcemia are Chovsteks and Trousseaus signs [31]. Patients may develop a prolonged QT on electrocardiogram [31]. Symptoms of hypocalcemia are not always evident at birth, with normal calcium and PTH levels however hypocalcemia evolves and unmasks over their life time [10,32]. Studies in knockout mice show that the appearance of PTH resistance is delayed and develops with age, despite the presence of the mutation since conception [32].
Hypocalcemia and hypophosphatemia, both of which are seen in the setting of PTH resistance, can be associated with the deposition of calcium phosphate products in extra-skeletal tissues. Calcium phosphate kidney stones, basal ganglia calcifications, band keratopathy, dental hypoplasia and soft tissue calcium-phosphate deposits have all been described in PHP [3,10,33]. PHP patients are at high risk for developing ectopic ossifications at birth or in early childhood [3], which are a manifestation of Gsα deficiency in mesenchymal stem cells, with de novo formation of extra-skeletal osteoblasts and the formation of ectopic bone islands in dermal and subcutaneous fat [34].
The second most common hormone resistance seen in PHP is TSH resistance, which manifests as the typical symptoms and signs of classic hypothyroidism. End organ resistance to TSH is seen in PHP 1A, PHP 1C and PHP 1B [3]. Other rare hormone abnormalities include gonadotropin resistance, Growth hormone deficiency [35] and hypercalcitoninemia [3,36].
The first large-scale epidemiological study evaluating morbidity and mortality in a PHP cohort was published 2016. Underbjerg L, et al. [4] reviewed the Danish National Patient Registry for patients with PHP and compared them to age and gender matched controls from the general background population. Compared to the control population, patients with PHP had an increased risk of infections (P < 0.01), cataracts (P < 0.01), seizures (P < 0.01) and neuropsychiatric disorders (P < 0.01). Furthermore, the risk of renal disorders, cardiovascular disorders, mortality, malignancy and fractures were comparable with the general background population.
iPPSD2 (PHP 1A and PHP 1C) [8]
Patients with PHP 1A and PHP 1C have features of AHO and multi-hormone resistance [10]. Patients with PHP 1A typically has short stature, early onset obesity, brachydactyly (70-80%), advanced bone age (70-80%), ectopic ossification (30-60%), as well as neurocognitive impairment (40-70%) [3]. PTH (100%), TSH (100%), partial gonadotropin resistance [37] and GHRH hormone resistance can be present with variable severity and present over a variable time course [6,38-40]. Females with PHP 1A develop hypogonadism manifested as delayed or incomplete sexual maturation, oligomenorrhea, amenorrhea or infertility [41]. GH deficiency related to resistance to GHRH has been reported in these patients [35,42].
Unlike patients with PHP 1A, patients with PHP 1C have normal Gsα bioactivity in red blood cells [43] but a blunted cAMP and phosphaturic response. Mutations in the last exon of GNAS (coding the receptor-interaction domain) have been detected in a few PHP 1C patients, suggesting that PHP 1C is an allelic variant of PHP 1A characterized by Gsα deficiency that selectively affects receptor coupling functions of Gsα [44].
iPPSD 2 (PPHP)
PPHP was originally described by Dr. Fuller Albright in 1952 [45]. Patients have the physical features of AHO, without the multi-hormonal resistance however some cases have mild resistance to PTH and TSH [22]. Patients typically have short stature, normal weight, brachydactyly (< 30%) and ectopic ossification (18-100%) [3].
iPPSD 3 (PHP 1B)
PHP 1B is characterized by renal resistance to PTH, in the absence of the typical features of AHO. Some patients may have one or more features of AHO, most commonly mild brachydactyly (15-33%) [3,46]. These patients typically achieve average adult height, have early onset obesity and advanced bone age (15-33%) [3]. TSH resistance can be seen in 30-100% patients with PHP 1B [3].
Management
The first international consensus statement on the diagnosis and management of PHP was published 6/2018 and will assist clinicians managing these rare disorders [3]. Patients with PHP, with PTH resistance, develop the clinical manifestations of hypocalcemia and hyperphosphatemia, which can be effectively managed with calcium supplements and activated vitamin D. Treatment of PHP should be directed at normalizing serum calcium and phosphate. The target PTH level is the upper end of the reference range to minimize the risk for long term hyperparathyroid related bone disease, [15] hypercalciuria and renal calcification [3].
It is recommended that patients have fasting albumin corrected calcium, serum PO4, Vitamin D 25OH, magnesium, PTH and creatinine values measured at baseline [47]. Asymptomatic hypocalcemia, may be treated with oral calcium supplements and if necessary, activated vitamin D, namely calcitriol. Activated vitamin D is preferred, over ergocalciferol or cholecalciferol, as it doesn't require PTH medicated 1-α-hydroxylation in the kidney, which is impaired in PHP disorders. In the acute setting, intravenous calcium gluconate and activated Vit D are necessary to raise the serum calcium [3]. It is necessary to monitor patient with telemetry during a calcium infusion [47].
Long-term, serum calcium levels are treated to a higher target than those for classic hypoparathyroidism, where the treatment target is the low normal range [31]. There are many different formulations of calcium, namely carbonate, citrate and phosphate. These calcium supplements can also function as phosphate binders in the gut lowering the serum phosphate level. Thiazide diuretics may be necessary to reduce urinary calcium losses, which are not typically high in PHP, in contrast to classic hypoparathyroidism [47]. Activated Vitamin D may be necessary to treat elevated PTH levels that are more than 2 times the upper level of normal, independent of the presence of hypocalcemia [3].
Patients with PHP, with PTH resistance, require regular monitoring of calcium, phosphate, vitamin D and PTH levels to avoid inadequate or excessive treatment of their disorder, specifically every 6 months in asymptomatic patients and more frequently when clinically indicated [3]. Calcium and phosphate homeostasis can fluctuate during acute illness, growth spurts and during pregnancy, necessitating more frequent monitoring of these electrolytes during these time periods [3,47]. Additionally, patients need to be educated on the symptoms and signs of both hypocalcemia and hypercalcemia [3].
Other hormone resistances associated with PHP are generally treated with hormone replacement. The second most common hormonal resistance is TSH resistance. This presents with the typical signs and symptoms of hypothyroidism. It is usually limited to patients with PHP 1A, PHP 1C and some patients with PHP 1B. Guidelines recommend TSH screening every 6 months in patients < 5years of age and yearly in older children and adults [3]. It may be necessary to evaluate the gonadotropin axis (follicle stimulating hormone, luteinizing hormone, estradiol, or testosterone) in cases of delayed puberty, infertility or amenorrhea, in PHP 1A and PHP 1C cases [3]. These patients may require exogenous estrogen or testosterone replacement. Evaluation of the GHRH-growth hormone axis (growth hormone, insulin-like growth factor-1) should also be considered in all patients with suspected PHP IA and PHP IC with poor growth velocity [3]. Children have been successfully treated with human GH replacement and have attained a successful height velocity in the pre-pubertal years [48]. Both children and adults with GH deficiency should be considered for GH replacement therapy [3].
The recent consensus guidelines on PHP recommend that patients have regular monitoring of BMI, eating behaviors, blood pressure, lipids and glucose levels, given the early onset of obesity (< 2 years of age) [3]. Sleep apnea is another recognized complication of obesity and patients should be screened with a detailed sleep history [3].
The current guidelines recommend imaging in patients with PHP if there is concern for AHO. Plain x-rays of the hands can easily identify shortened metacarpals. Chronic hypocalcemia and hyperphosphatemia, related to untreated PHP, can result in intracranial deposits of calcium, also known as Fahr syndrome [31]. Basal ganglia imaging with a plain skull x-ray or CT head may be necessary in patients with movement disorders or to exclude other causes of paresthesias or seizures [3]. Patients may need an ophthalmologic evaluation to evaluate for cataract development [3]. Patients with PHP are at risk for bone loss as there is preserved sensitivity to PTH at the level of the bone, necessitating a need for a bone density scan if there are other risk factors for osteoporosis such as hypogonadism or gonadotropin resistance, GH deficiency or the postmenopausal status [3].
Management of patients with PHP is complex and involves life-long management of multi-hormone resistances, metabolic and skeletal complications associated with this rare disorder. Mutational analysis of the GNAS gene that identifies PHP 1A, PHP IB and PPHP is available and may be necessary to definitively diagnose challenging PHP cases. It is important to make a diagnosis for PHP and thus treat these patients appropriately to reduce the severity and complications of the disease.
Conclusions
PHP encompasses a spectrum of orphan disorders with variable degree of hormone resistance and skeletal manifestations leading to a limited experience in managing these patients by many clinicians. These patients need life-long follow-up for metabolic complications, as well as the development of hormone resistances over time. The publication of the first international consensus statement on the diagnosis and management of PHP is an important contribution to the future management of these patients. It is important to establish a correct diagnosis of PHP to enable appropriate treatment and genetic counselling of these individuals and their first-degree relatives.
I do not have any conflicts of interest to report.
References
- Albright F, Burnett CH, Smith CH, et al. (1942) Pseudohypoparathyroidism: An example of "Seabright-Bantam syndrome". Endocrinology 30: 922-932.
- Breslau NA, Weinstock RS (1988) Regulation of 1,25OH2D synthesis in hypoparathyroidism and pseudohypoparathyroidism. Am J Physiol 255: E730-E736.
- Mantovani G, Bastepe M, Monk D, et al. (2018) Diagnosis and management of pseudohypoparathyroidism and related disorders: First international consensus statement. Nat Rev Endocrinol 14: 476-500.
- Underbjerg L, Sikjaer T, Mosekilde, et al. (2016) Pseudohypoparathyroidism-epidemiology, mortality and risk of complications. Clin Endocrinol (Oxf) 84: 904-911.
- Nakamura Y, Matsumoto T, Tamakoshi A, et al. (2000) Prevalence of idiopathic hypoparathyroidism and pseudohypoparathyroidism in Japan. J Epidemiol 10: 29-33.
- Levine MA, Downs RW, Moses AM, et al. (1983) Resistance to multiple hormones in patients with pseudohypoparathyroidism: Association with deficient activity of guanine nucleotide regulatory protein. Am J Med 74: 545-556.
- Farfel Z, Brickman AS, Kaslow HR, et al. (1980) Defect of receptor-cyclase coupling protein in pseudohypoparathyroidism. N Engl J Med 303: 237-242.
- Thiele S, Mantovani G, Barlier A, et al. (2016) From pseudohypoparathyroidism to inactivating PTH/PTHrP signaling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol 175: 1-17.
- Elli FM, Linglart A, Garin I, et al. (2016) The prevalence of GNAS deficiency-related disease in a large cohort of patients characterized by the EuroPHP network. J Clin Endocrinol Metab 101: 3657-3668.
- Mantovani G (2011) Clinical review: Pseudohypoparathyroidism: Diagnosis and treatment. J Clin Endocrinol Metab 96: 3020-3030.
- Kuzel AR, Lodhi MU, Rahim M (2017) Classic and non-classic features in pseudohypoparathyroidism: Case study and brief literature review. Cureus 9: e1878.
- Pena C, Pinochet C, Florenzano P, et al. (2018) Pseudohypoparathyroidism: Report of two cases of late presentation. Rev Med Chil 146: 116-121.
- Juppner H, Bastepe M (2013) Pseudohypoparathyroidism. In: CJ Rosen, Primer on the metabolic bone diseases and disorders of mineral metabolism. (8th edn), 73: 590-600.
- Stone M, Hosking D, Garcia-Himmelstine C, et al. (1993) The renal response to exogenous parathyroid hormone in treated pseudohypoparathyroidism. Bone 14: 727-735.
- Murray T, Gomez Rao E, Wong MM, et al. (1993) Pseudohypoparathyroidism with osteitis fibrosa cystica: Direct demonstration of skeletal responsiveness to parathyroid hormone in cells cultured from bone. J Bone Miner Res 8: 83-91.
- Tollin SR, Perimutter S, Alola JF (2000) Serial changes in bone mineral density and bone turnover after correction of secondary hyperparathyroidism in a patient with pseudohypoparathyroidism type 1B. J Bone Miner Res 15: 1412-1416.
- Chase LR, Melson GL, Aurbach GD (1969) Pseudohypoparathryoidism: Defective excretion of 3',5'-AMP in response to parathyroid hormone. J Clin Invest 48: 1832-1844.
- Chong PL, Meeking DR (2013) Pseudohypoparathyroidism: A rare but important cause of hypocalcemia. BMJ Case Rep.
- Seki T, Yamamoto M, Kumura H, et al. (2010) Vitamin D deficiency in two young adults with biochemical findings resembling pseudohypoparathyroidism type 1 and type 2. Endocr J 57: 735-744.
- Todorova-Koteva K, Wood K, Imam S, et al. (2012) Screening for parathyroid hormone resistance in patients with nonphenotypically evident pseudohypoparathyroidism. Endocr Pract 18: 864-869.
- Epstein M, McGrath S, S Law (2006) Proton pump inhibitors and hypomagnesemia hypoparathyroidism. N Engl J Med 355: 1834-1836.
- Turan S, Thiele S, Tafaj O, et al. (2015) Evidence of hormone resistance in a pseudo-pseudohypoparathyroid patient with a novel paternal mutation in GNAS. Bone 71: 53-57.
- Tafaj O, Juppner H (2017) Pseudohypoparathyroidism: One gene, several syndromes. J Endocrinol Invest 40: 347-356.
- Long DN, McGuire S, Levine MA, et al. (2007) Body mass index differences in pseudohypoparathyroidism type 1A versus pseudopseudohypoparathyroidism may implicate paternal imprinting of Galpha (s) in the development of human obesity. J Clin Endocrinol Metab 92: 1073-1079.
- Levine MA, Jap TS, Mauseth RS, et al. (1986) Activity of the stimulatory guanine nucleotide-binding protein is reduced in erythrocytes from patients with pseudohypoparathyroidism and pseudopseudohypoparathyroidism: Biochemical, endocrine, and genetic analysis of Albrights hereditary osteodystrophy in six kindreds. J Clin Endocrinol Metab 62: 497-502.
- Zazo C, Thiele S, Martín C, et al. (2011) Gsα activity is reduced in erythrocyte membranes of patients with psedohypoparathyroidism due to epigenetic alterations at the GNAS locus. J Bone Miner Res 26: 1864-1870.
- Davies SJ, Hughes HE (1993) Imprinting in Albrights hereditary osteodystrophy. J Med Genet 30: 101-103.
- Farfel Z, Bourne HR, Iiri T (1999) The expanding spectrum of G protein diseases. N Engl J Med 340: 1012-1020.
- De Sanctis L, Vai S, Andreo MR, et al. (2004) Brachydactyly in 14 genetically characterized pseudohypoparathyroidism type 1a patients. J Clin Endocrinol Metab 89: 1650-1655.
- Chen M, Wang J, Dickerson KE, et al. (2009) Central nervous system imprinting of the G protein G(s)alpha and its role in metabolic regulation. Cell Metab 9: 548-555.
- Shoback D, Bilezikian JP, Costa AG, et al. (2016) Presentation of hypoparathyroidism: Etiologies and clinical features. J Clin Endocrinol Metab 101: 2300-2312.
- Turan S, Fernandez-Rebollo E, Aydin C, et al. (2014) Postnatal establishment of allelic Gαs silencing as a plausible explanation for the delayed onset of parathyroid hormone resistance owing to heterozygous Gαs disruption. J Bone Miner Res 29: 749-760.
- Stieler K, Schnabel D, Atugoda S, et al. (2011) Albright hereditary osteodystrophy. Pediatr Dermatol 28: 135-137.
- Pignolo RJ, Xu M, Russel E, et al. (2011) Heterozygous inactivation of the GNAS in adipose-derived mesenchymal progenitor cells enhances osteoblast differentiation and promotes heterotopic ossification. J Bone Miner Res 26: 2647-2655.
- Germain-Lee EL, Groman J, Crane JL, et al. (2003) Growth hormone deficiency in pseudohypoparathyroidism type 1A: Another manifestation of multihormone resistance. J Clin Endocrinol Metab 88: 4059-4069.
- Vlaeminck-Guillem V, D'Herbomez M, Pigny P, et al. (2001) Pseudohypoparathyroidism 1A and hypercalcitoninemia. J Clin Endocrinol Metab 86: 3091-3096.
- Namnoum AB, Merriam GR, Moses AM, et al. (1998) Reproductive dysfunction in women with Albright's hereditary osteodystrophy. J Clin Endocrinol Metab 83: 824-829.
- Weinstein LS, Yu S, Warner DR, et al. (2001) Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 22: 675-705.
- Wemeau JL, Balavoine AS, Ladsous M, et al. (2006) Multihormonal resistance to parathyroid hormone, thyroid stimulating hormone, and other hormonal and neurosecretory stimuli in patients with pseudohypoparathyroidism. J Pediatr Endocrinol Metab 19: 653-661.
- Germain-Lee EL (2006) Short stature, obesity, and growth hormone deficiency in pseudohypoparathyroidism type 1A. Pediatr Endocrinol Rev 3: 318-327.
- Mantovani G, Spada A (2006) Resistance to growth hormone releasing hormone and gonadotropins in Albright's hereditary osteodystrophy. J Pediatr Endocrinol Metab 19: 663-670.
- DeSanctis L, Bellone J, Salerno M, et al. (2007) GH secretion in a cohort of children with pseudohypoparathyroidism type 1A. Journal of Endocrinological Investigation 30: 97-103.
- Farfel Z, Brothers WM, Brickman AS, et al. (1981) Pseudohypoparathyroidism: Inheritance of deficient receptor-cyclase coupling activity. Proc Natl Acad Sci USA 78: 3098-3102.
- Thiele S, de Sanctis L, Werner R, et al. (2011) Functional characterization of GNAS mutations found in patients with pseudohypoparathyroidism type 1C defines a new subgroup of pseudohypoparathyroidism affecting selectively Gsα-receptor interaction. Hum Mutat 32: 653-660.
- Albright F, Forbes AP, Henneman PH (1952) Pseudopseudohypoparathyroidism. Trans Assoc Am Physicians 65: 337-350.
- Mantovani G, de Sanctis L, Barbieri AM, et al. (2010) Pseudohypoparathyroidism and GNAS epigenetic defects: clinical evaluation of Albright hereditary osteodystrophy and molecular analysis in 40 patients. J Clin Endocrinol Metab 95: 651-658.
- Gittoes N, Ayuk J. Pseudohypoparathyroidism. BMJ best practice online. Accessed 7/10/18
- Mantovani G, Ferrante E, Giavoli C, et al. (2010) Recombinant human GH replacement therapy in children with pseudohypoparathyroidism type IA: first study on the effect on growth. J Clin Endocrinol Metab 95: 5011-5017.
- Romanet P, Osei L, Netchine I, et al. (2015) A case report of GNAS epigenetic defect revealed by a congenital hypothyroidism. Pediatrics 135: e1079-e1083.
- Kayemba-Kays S, Tripon C, Heron A, et al. (2016) Pseudohypoparathyroidism Type 1A - subclinical hypothyroidism and rapid weight gain as early clinical signs: a clinical review of 10 cases. J Clin Res Pediatr Endocrinol 8: 432-438.
- Fernández-Rebollo E, Lecumberri B, Gaztambide S, et al. (2013) Endocrine profile and phenotype-(Epi)genotype correlation in spanish patients with pseudohypoparathyroidism. J Clin Endocrinol Metab 98: 996-1006.
Corresponding Author
Bridget P Sinnott, Associate Professor of Medicine, Division of Diabetes, Endocrinology and Metabolism, Medical College of Georgia, 1447 Harper St, HB-5025, Augusta, GA 30912, USA, Tel: 706-721-2131, Fax: 706-721-6892.
Copyright
© 2018 Sinnott BP. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.