Endothelial Cell Activation Mediates Platelet Response to Pulsatile Shear Stress
Abstract
Thrombin plays critical roles in both hemostasis and thrombosis. Many studies were carried out to examine the effects of different thrombogenic surfaces on thrombin generation. In the present study, how activated endothelial cells affected platelet thrombin generation under dynamic shear stress was investigated. Human platelets were exposed to pulsatile shear stress (physiological, low or elevated) in a cone-and-plate shearing device for 30 minutes in the presence of quiescent or activated human coronary artery endothelial cells. Coagulation factors including Factor II, Factor Xa and calcium were immediately available. Thrombin generation rates were measured using a modified prothrombinase assay. Platelet expression of surface P- selectin, glycoprotein Ibα (GPIbα or CD42b), integrin αIIb (CD41a), and phosphatidylserine was measured using flow cytometry. Endothelial cell expression of intracellular adhesion molecule 1 (ICAM-1) and von Willebrand factor was measured using a solid-phase ELISA and fluorescence microscopy, respectively. Platelet adhesion to the endothelial cells was measured using fluorescence microscopy. The results indicated that the activation state of endothelial cells, but not the pulsatile shear stress, had a significant effect in thrombin generation rates. The activation state of endothelial cells also affected platelet surface GPIbα and P-selectin expression, which may have contributed to the overall change in thrombin generation rates.
Keywords
Platelets, Shear stress, Endothelial cells, Thrombin generation
Introduction
Vascular injury triggers a complex weave of cellular activation, intracellular signalling, and biochemical reactions collectively termed hemostasis. The overall goal of hemostasis is to prevent blood loss at the site of vascular insult. A key component of hemostasis is the coagulation cascade, in which numerous blood coagulation factors are activated, ultimately resulting in the formation of fibrin. Circulating platelets are key players in multiple stages of hemostasis, including thrombin generation [1]. Platelets contribute to the formation of the prothrombinase complex, which cleaves prothrombin (Factor II) into its activated form, thrombin (Factor IIa). Activated platelets release activated coagulation Factor V (Va), which binds to Factor Xa to form the prothrombinase complex. Platelets also, through an increased expression of phosphatidylserine (PS) on the outer membrane, provide a negatively charged surface that enables the enzymatic reaction of prothrombin cleavage [2].
Thrombin is a critically important protein with multiple functions in coagulation. It converts soluble plasma protein fibrinogen into insoluble fibrin and activates coagulation Factor XIII. Activated Factor XIII, i.e., FXIIIa, cross-links the fibrin polymers into a tough mesh that can trap circulating blood cells to form a clot. Rate of thrombin generation in a given patient can have clinical implications. Slow generation of thrombin in the initial stages of coagulation can result in a clot that is susceptible to dissolution via fibrinolysis, whereas rapid thrombin generation yields a much more stable clot [3].
To better understand thrombin generation kinetics, many models (computational, in vitro and in vivo) have been developed to investigate factors that have significant impact on thrombin generation rate. A study by Diquélou, et al. found that the type of thrombogenic surface had an impact on platelet-dependent thrombin generation [4]. They reported that at different shear rates (50, 650, and 2600 s-1), procoagulant endothelial cells and collagen type III induced similar levels of thrombin generation. It was found that the efficacy of antiplatelet therapies is dependent on shear rate [5]. Clinical observations showed that aspirin (an irreversible thromboxane A2 inhibitor) was antithrombotic in mild stenosis (where shear rate/stress is modestly increased), but ineffective as an antithrombotic drug for severe stenosis (exceeding 90% occlusion, where shear rate/stress is greatly increased) [6]. This was in agreement with an in vitro study by Barstad et al., which demonstrated platelet aspirin insensitivity at high shear rates (10,500 s-1) [7]. Adherent tissue factor (TF) also has a shear-dependent effect on platelet adhesion and activation. Tonda et al. [8] found that platelet adhesion increased as shear rate decreased from 600 s-1 to 250 s-1, as did fibrin formation. This increase in fibrin formation has also been shown to be concentration-dependent, with a threshold concentration of 2-10 molecules of TF/μm2 required to trigger a significant response at both venous (100 s-1) and arterial (1000 s-1) shear rates [9-10]. Similar findings over collagen [11-13] and von Willebrand Factor [14-15] have been reported, the latter being critical to platelet adhesion under arterial flow conditions (where physiological wall shear rates are about 1000 s-1-2000 s-1 and stenotic vessel wall shear rates can exceed 5000 s-1) [16]. Ex vivo models utilizing denuded animal arteries have also been used to test thrombin generation kinetics in the flow regime. A 2017 study found that the anticoagulant drug apixaban (a Factor Xa inhibitor) inhibited platelet-mediated thrombin generation in a dose-dependent manner under static conditions, but at a shear rate of 800 s-1, only the highest dose of the drug (160 ng/mL) was sufficient to prevent platelet and fibrin aggregates (17).
Most of these previous studies were performed using a wide range of shear rates, from 50 s-1 to over 10000 s-1. However, these shear rates were applied constantly over the duration of the studies. We reported previously that shear stress patterns (i.e., constant or pulsatile) and the availability of coagulation factors can also affect thrombin generation rates [18]. Additionally, the interaction between platelets and vascular wall endothelial cells could have a significant impact on platelet behaviour [19-20]. Healthy endothelial cells can release platelet activation inhibiting vasodilators such as nitric oxide and prostacyclin, while injured endothelium can expose higher levels of tissue factor, triggering the extrinsic pathway coagulation [21-22]. Activated endothelial cells also have enhanced vWF and E-selectin expression, which can promote platelet adhesion and rolling [22-24]. However, how endothelial cells affect thrombin generation rates under dynamic flow conditions is not well understood. In the current study we aim to fill this gap. We hypothesized that endothelial cell activation state would affect platelet thrombin generation rates under dynamic flow conditions. To test this hypothesis, human platelets were exposed to pulsatile shear stress in the presence of human coronary artery endothelial cells (quiescent or activated); thrombin generation rate, platelet activation, platelet surface adhesion protein expression, platelet adhesion to endothelial cells, as well as endothelial cell activation and surface adhesion protein (von Willebrand factor) expression were measured.
Methods
Washed platelets preparation
Fresh platelet-rich plasma (PRP) from healthy donors was purchased from Oklahoma Blood Institute (Oklahoma City, Oklahoma) [18-19]. Washed platelets were prepared from centrifugation of PRP at 1000 × g followed by resuspension in platelet buffer (135 mM NaCl, 5 mM Glucose, 2.7 mM KCl, 0.5 mM Na2HPO4, 1 mM MgCl2, 1 mM sodium citrate, 0.1% bovine serum albumin, and 10 mM HEPES, pH 7.4 [25]. Final platelet concentration was 100,000 platelets/μL [26].
Endothelial cell culture
Human coronary artery endothelial cells (HCAECs) were purchased from ScienCell Research Laboratories (Carlsbad, California). Cells were grown in a monolayer to confluence on 6-well plates pre-coated with 2 μg/mL fibronectin (Sigma Aldrich). HCAECs were maintained in endothelial cell media supplemented with 5% fetal bovine serum (FBS), endothelial cell growth supplements (ECGS), and penicillin/streptomycin. All cell culture reagents were purchased from ScienCell Research Laboratories. Cells were used between passages 2 and 7. To activate endothelial cells, HCAECs were treated with lipopolysaccharide (LPS, 1μg/mL, Sigma Aldrich) for up to 48 hours prior to the experiments. Prior to shearing, HCAECs were washed twice with PBS to remove the media and LPS.
Shear stress treatment
Washed platelets (100,000 /μL) were exposed to pulsatile shear stress in a cone-and-plate shearing device (based on a cone-and-plate viscometer and made in house [27]) for 30 minutes [18] in the presence of cultured endothelial cells (ECs). In some experiments, endothelial cells were activated using LPS. Three pulsatile shear waveforms mimicking low, normal, and elevated shear stress in the coronary artery were used to stimulate platelets [19,28]. In a healthy artery, shear stresses range between 0-1 Pa over the cardiac cycle ("normal" shear condition). With the presence of a 70% stenosis, shear stresses in the narrowed region of the artery (or stenosis throat) can reach 3 Pa ("stenosis" shear condition). When platelets travel out of the narrowed region, they will experience normal shear stress (0-1 Pa) again. Downstream of the stenosis throat, within a recirculation zone, shear stress is low, ranging from 0-0.3 Pa near the vessel wall ("recirculation" shear condition).
Thrombin generation rate
We previously reported that the availability of coagulation factors played an important role in thrombin generation rates [18]. In this study, when platelets were exposed to shear stress, Ca2+ (5 mM), Prothrombin (Factor II, 1.41 μM), and Factor Xa (0.1 nM) were provided at the same time in the cone and plate shearing device.
Following shear stress treatment, 10 μL of platelet sample was removed from the cone and plate shearing device and directly added to a 96-well plate containing Chromozym-Th (Roche Ltd). Color development was read at 405 nm in a microplate reader (Molecular Devices). Thrombin generation was calculated using a standard curve, and thrombin generation rate was calculated as the slope of thrombin concentration over time. Platelets that were not exposed to shear stress or ECs (i.e., resting platelets) were used as the experimental control.
Platelet activation and surface adhesion protein expression
Platelets were incubated with fluorescent antibodies for 30 minutes at room temperature in the dark, followed by flow cytometry (Accuri C6, BD Biosciences) immediately. Platelet surface P-selectin expression was measured using a FITC-conjugated murine anti human CD62P antibody (Ancell Corporation, 1:50 dilution). Platelet surface GPIb expression was measured using a FITC-conjugated murine anti-human CD42b antibody (BD Pharminigen, 1:50 dilution). Platelet surface αIIb expression was measured using a FITC-conjugated murine anti-human CD41a antibody (Ancell Corporation, 1:50 dilution). Platelet surface phosphatidylserine (PS) expression was measured using FITC-conjugated Annexin V (Life Technologies, 1:50 dilution). Non-specific antibody binding was measured using a FITC-conjugated mouse anti-human IgG antibody (BD Pharminigen, 1: 50 dilution). Platelet population was gated based on the forward and side scatter in a population density plot. Gating of the positive and negative populations were set relative to the non-specific mouse IgG antibody fluorescence intensity histograms.
Platelet adhesion and endothelial surface von Willebrand Factor expression
Following shear stress treatment, supernatant containing platelets were removed from the plates. After washing with PBS (phosphate buffered saline, PH 7.4), cells (platelets that adhered to the bottom of the plate, and ECs) were fixed using 0.5% glutaraldehyde, followed by neutralization with 100 mM glycine-BSA (Sigma Aldrich). Alexa Fluor 488-conjugated phalloidin was used to detect platelet/EC cytoskeleton F actin filaments. Alexa Fluor 647-conjugated murine anti-human von Willebrand Factor (vWF) antibody was used to detect endothelial cell surface vWF expression, and DAPI was used to detect endothelial cell nuclei. Immunofluorescence microscopy was performed to visualize platelet adhesion to endothelial cell monolayer. Using the Image J software, the number of platelets adhered to the EC monolayer was counted. Endothelial cell surface vWF expression was quantified using the mean fluorescence intensity and normalized to quiescent ECs.
Endothelial cell activation
EC activation was assessed using a solid phase ELISA approach, by measuring ICAM-1 expression. As described before, following shear stress treatment with platelets, ECs were washed and fixed. Cell surface ICAM-1 expression was measured using a murine anti-human ICAM-1 antibody (Abcam). Primary antibody binding was detected using an alkaline phosphatase-conjugated goat anti mouse secondary antibody and a pNPP (p-nitrophenyl phosphate) substrate.
Statisics
Data was analyzed using Student's t test or two-way ANOVA in SAS (v. 9.3). The two factors were shear stress conditions (recirculation, normal or stenosis) and EC activation state (quiescent or activated). When significant difference (P < 0.05) was detected, the Least Square Means (LSM) post hoc test was used for further comparison.
Results
Endothelial cell activation
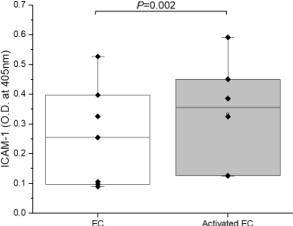
EC activation was confirmed by ICAM-1 expression. Figure 1 demonstrates that following LPS treatment (1μg/mL), EC surface ICAM-1 expression increased by 30% from 0.256 ± 0.038 (mean ± standard error, n = 5) to 0.334 ± 0.075 (n = 7). Normality of the data was confirmed with the Shapiro-Wilk test (P = 0.177), and paired Student's t-test indicated that the difference was significant (P = 0.002), confirming LPS-treated ECs were activated.
Thrombin generation rate
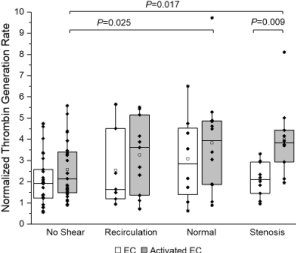
Figure 2 depicts normalized platelet thrombin generation rates in the presence of ECs (quiescent and activated). All thrombin generation rates were normalized (as fold change) to that of quiescent (control) platelets from the same day. With the presence of LPS-activated ECs, platelets exposed to normal and stenosis shear stress had a significant increase (3.83 ± 0.70, n = 12, P = 0.025 for normal condition and 4.01 ± 0.58, n =10, P = 0.017 for stenosis condition, respectively) in normalized thrombin generation rates compared to platelets that were not exposed to shear stress (2.57 ± 0.26, n = 27) (Figure 2). However, with the presence of quiescent ECs, no significant differences were observed under normal or stenosis shear stress conditions. For platelets exposed to stenosis shear stress, the presence of LPS-activated ECs induced a significant increase (P = 0.009) in normalized platelet thrombin generation rate (4.01 ± 0.58, n = 10), compared to when quiescent ECs were present (2.11 ± 0.80, n = 10). Two-way ANOVA indicated that the activation state of ECs had a significant (P = 0.005) impact on thrombin generation rates, while overall, 30 min shear stress treatment (recirculation, normal or stenosis) did not (P > 0.05).
Platelet surface activation/adhesion protein Expression
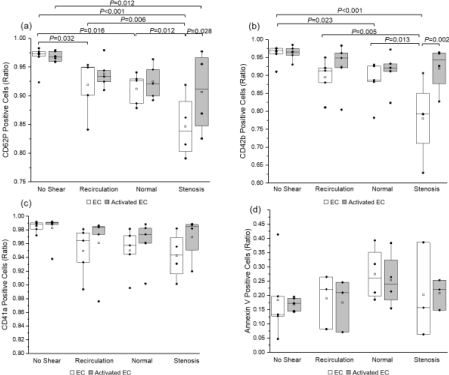
Platelet surface expression of activation (P-selectin or CD62P) and adhesion proteins (GPIbα or CD42b, which binds to von Willebrand Factor; and integrin αIIb or CD41a, part of the αIIbβIII which is a receptor for both vWF and fibrinogen) is depicted in (Figure 3). Two-way ANOVA indicated that platelet surface CD62P expression was not significantly affected by EC activation state. However, increasing shear stress led to a reduction in the number of platelets positive for P-selectin (Figure 3a). For platelets sheared over quiescent ECs, the percentage of platelets positive for CD62P decreased from 96.7% ± 0.8% under no shear conditions (n = 7) to 91.9% ± 2.2% (n = 5, P = 0.032) under recirculation conditions, 91.2% ± 1.2% (n = 5, P = 0.016) under normal conditions, and 84.7% ± 2.8% (n = 4, P < 0.001) under stenosis conditions. No differences were observed between ECs and activated ECs at all magnitudes except under stenosis conditions. Overall, shear stress magnitude had a significant impact on CD62P expression (P < 0.001), while EC activation state did not (P = 0.057). In comparison, both shear stress (P = 0.001) and EC activation state (P = 0.009) caused significant changes in platelet surface GPIbα expression (Figure 3b).
In the presence of quiescent ECs, the percentage of CD42b-positive platelets decreased significantly under normal shear stress (88.2% ± 2.7%, n = 5) compared to no shear stress (96.2% ± 0.9%, n = 7, P = 0.023); stenosis shear stress further decreased the percentage of CD42b-positive platelets (78% ± 5.7%, n = 4, P = 0.013 when compared to normal shear, and P < 0.001 when compared to no shear). Under stenosis shear stress, the percentage of CD42b-postive platelets sheared over LPS-activated ECs (92.0% ± 3.2%, n = 4) was significantly greater than platelets sheared over quiescent ECs (78.0% ± 5.7%, n = 4, P = 0.002). By contrast, integrin αIIb expression (Figure 3c) and phosphatidylserine expression (Figure 3d) was not significantly impacted by either EC activation state (P = 0.343 and P = 0.775, respectively) or shear stress magnitude (P = 0.070 and P = 0.319 respectively).
Von willebrand factor expression
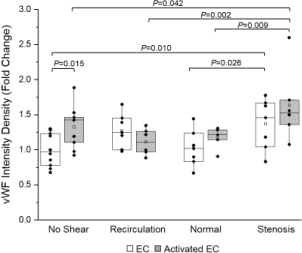
Figure 4 depicts von Willebrand Factor (vWF) expression on ECs exposed to shear stress with platelets and coagulation factors. Quiescent ECs showed increased vWF expression under high pulsatile shear stress mimicking stenosis conditions (1.37 ± 0.13, n = 7) compared to normal shear stress (1.02 ± 0.09, n = 7, P = 0.026). LPS-activated ECs showed a similar pattern in vWF expression, with significant differences between high (1.63 ± 0.21, n = 6), low (1.11 ± 0.08, n = 6, P = 0.002), and normal shear stress (1.17 ± 0.07, n = 5, P = 0.009). When there was no shear stress, activated ECs showed significantly increased expression of vWF compared to quiescent ECs (P = 0.015). However, this pattern did not hold over all shear stress conditions (P = 0.065).
Platelet adhesion
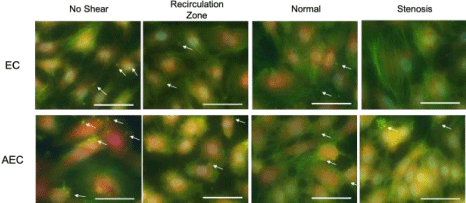
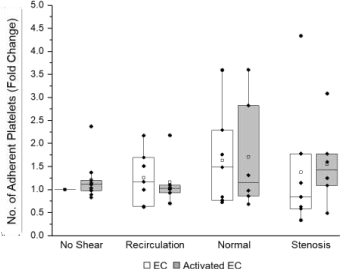
Representative image of platelet adhesion to ECs are shown in (Figure 5). Alexa Fluor 488-conjugated phalloidin phalloidin (which binds to cytoskeleton F actin filaments) was used to label both platelets and ECs (green fluorescence); and an Alexa Fluor 647-conjugated vWF antibody (red fluorescence) and DAPI (blue) were used to detect ECs. The normalized (to the area of ECs) numbers of adhered platelets are depicted in (Figure 6). More platelets appeared to adhere to ECs (quiescent or activated) under normal shear stress, compared to recirculation or stenosis shear stress. However, no significant differences were detected in the number of platelets adhered to inactivated and LPS-activated ECs, and among different shear stress conditions (no shear, recirculation, normal or stenosis).
Discussion
Platelet activation and thrombin generation under various shear stress conditions and over various substrates have been extensively studied (29-31). Compared to those reports, the physiological relevance of the current study was significantly improved, as pulsatile shear stress mimicking that in the coronary arteries was used, coagulation factors were provided simultaneously when platelets were exposed to shear flow, and the activation state of ECs was considered. Washed platelets but not whole blood or plasma (both contain many proteins/factors that can enhance or inhibit coagulation) were used in this study, so measured platelet responses were directly resulted from shear stress and endothelial cells. The study demonstrated that the activation state of ECs had a more significant impact (than shear stress patterns) on thrombin generation rates (Figure 2).
In our previous report [18], we demonstrated that when the coagulation factors were immediately available, platelet thrombin generation rate did not increase with shear stress (or shear rate) magnitude; instead, elevated (stenosis) shear stress caused the lowest thrombin generation following platelet shear exposure. We suspected that it was due to the short residence time of coagulation factors (in contact with platelets) under shear flow. With the presence of quiescent ECs, a similar trend was observed, i.e., platelets had a lower thrombin generation rate under stenosis shear stress conditions. However, with the presence of activated ECs, platelets exposed to stenosis (0-3 Pa) shear stress had the highest rate of thrombin generation within 30 minutes (Figure 2), suggesting that EC activation could enhance shear stress-induced platelet activation and thrombin generation, or EC activation could increase the residence time (possibly by slowing down or immobilizing platelets) under high shear flow.
Expression of platelet and EC surface adhesion molecules was dependent on both shear stress conditions and EC activation state. Stenosis shear stress (0-3 Pa) caused a significant increase in EC vWF expression in both quiescent and activated ECs (Figure 4), which is consistent with previously reported data [32, 33]. Contrary to what we observed before under constant shear stress conditions (18), platelet surface P-selectin expression decreased as shear stress magnitude increased (especially when quiescent ECs were present). The presence of activated ECs caused a significance increase (P = 0.028) in platelet surface P-selectin expression under stenosis shear stress conditions. GPIbα (CD42b) expression was universally higher in platelets sheared over activated ECs compared to quiescent ECs; but shear stress magnitudes did not seem to have an impact on GPIbα (CD42b) expression when ECs were activated.
When quiescent ECs were present, however, GPIbα expression decreased significantly as shear stress magnitude increased. This suggests platelets can activate under low pulsatile shear stress, especially when they are in direct contact with activated ECs. Platelet surface phosphatidylserine expression was measured by Annexin V inding using flow cytometry. The results indicated that there was no significant difference in phosphatidylserine expression between different shear stress conditions. This is similar to what we observed previously, when platelets were exposed to pulsatile shear stress without the presence of endothelial cells [18].
As depicted in Figure 6, neither shear stress nor EC activation state caused any significant changes in the number of platelets adhered to ECs (quiescent or activated). There may be several reasons for this. In this study, washed platelets instead of whole blood or platelet-rich plasma (PRP) were used so that thrombin generation would only be impacted by EC activation state and/or shear stress, but not other blood components. The removal of plasma proteins that participate in the adhesion and aggregation of platelets, such as fibrinogen and soluble vWF, may have affected the adhesion we observed here. Second, platelet GPIbα expression and EC vWF expression displayed opposite behavior in response to shear stress, the former being significantly reduced while the latter significantly increased when shear stress increased. It is possible that the relative scarcity of one at one shear stress magnitude may cancel out the relative abundance of the other, leading to similar levels of platelet adhesion. Third, while GPIbα-vWF bonds are considered to be crucial in arterial thrombosis due to their fast on-rate (34,35), they are relatively transient compared to the bonds between other surface proteins (such as GPVI and αIIbβIII) to their ligands (36). The fact that adhesion neither increased nor decreased despite variations in GPIbα and/or vWF expression may indicate that these bonds have less of an impact on sustained adhesion under dynamic flow conditions. Lending credence to this idea, we observed no difference in αIIb expression in our experiments (Figure 3c). In this study, washed platelets were used. Since no plasma FV/FVa was involved, FVa that contributed to prothrombinase complex formation and thrombin generation was provided solely by activated platelets. Therefore, the amount of FVa released by activated platelets was likely the major factor that determined thrombin generation rates under different shear stress and EC activation conditions. To further improve the physiological relevance of this in vitro thrombin generation model, fibrinogen or soluble vWF may need to be added to the platelet-coagulation factor mixture in the cone and plate shearing device to investigate what, if any, effect these proteins may have on thrombin generation and platelet activation. Additionally, while we elected to use a cone-and-plate shearing device to apply spatially uniform shear stress to platelets and endothelial cells, a channel system (37,38) might better replicate the flow patterns in a blood vessel, although this method presents challenges in the imaging of ECs and platelet adhesion.
In summary, we examined the response of platelets and endothelial cells when they were exposed to physiologically relevant dynamic flow conditions together. EC activation state could significantly affect platelet thrombin generation rate, especially under normal and stenosis shear stress conditions. EC activation state can also affect platelet surface adhesion protein, i.e., P-selection and GPIbα (CD42b) expression under shear flow. Therefore, EC activation state must be taken into consideration when platelet thrombin generation kinetics is evaluated under flow.
Acknowledgements
The authors would like to thank Srividya Alisetti, Sarah Choudhury, and Yu-Xuan Li for their help with the analysis of adhesion data.
Statement of Ethics
Human plasma was purchased from Oklahoma Blood Institute, which acquired informed consent from all donors. All plasma samples were de-identified prior to shipment. No animal work was conducted for this study.
Conflict of Interest Statement
The authors report no conflicts of interest.
Funding
This work was supported in part by an American Heart Association Pre-doctoral Fellowship Award (ES,19PRE34450210), and an American Heart Association Grant in Aid Award (WY, 16GRNT30440004).
References
- Hvas AM (2016) Platelet function in thrombosis and hemostasis. Semin Thromb Hemost 42: 183-184.
- Vance JE, Steenbergen R (2005) Metabolism and functions of phosphatidylserine. Prog Lipid Res 44: 207-234.
- Wolberg AS, Allen GA, Monroe DM, et al. (2005) High dose factor VIIa improves clot structure and stability in a model of haemophilia B. Br J Haematol 131: 645-655.
- Diquélou A, Lemozy S, Dupouy D, et al. (1994) Effect of blood flow on thrombin generation is dependent on the nature of the thrombogenic surface. Blood 84: 2206-2213.
- Hanson SR, Sakariassen KS (1998) Blood flow and antithrombotic drug effects. Am Heart J 135: 132-145.
- Veen G, Meyer A, Verheugt FW, et al. (1993) Culprit lesion morphology and stenosis severity in the prediction of reocclusion after coronary thrombolysis: Angiographic results of the APRICOT study. J Am Coll Cardiol 22: 1755-1762.
- Barstad RM, Orvim U, Hamers MJ, et al. (1996) Reduced effect of aspirin on thrombus formation at high shear and disturbed laminar blood flow. Thromb Haemost 75: 827-832.
- Tonda R, Lopez-Vilchez I, Navalon F, et al. (2008) Platelets interact with tissue factor immobilized on surfaces: Effects of shear rate. Eur J Clin Invest 38: 34-42.
- Okorie UM, Denney WS, Chatterjee MS, et al. (2008) Determination of surface tissue factor thresholds that trigger coagulation at venous and arterial shear rates: Amplification of 100 fM circulating tissue factor requires flow. Blood 1: 3507-3513.
- Diamond SL (2010) Tissue factor activity under flow. Thromb Res 125: S29-S30.
- Li R, Fries S, Li X, et al. (2013) Microfluidic assay of platelet deposition on collagen by perfusion of whole blood from healthy individuals taking aspirin. Clin Chem 59: 1195-1204.
- Zhu S, Tomaiuolo M, Diamond SL (2016) Minimum wound size for clotting: Flowing blood coagulates on a single collagen fiber presenting tissue factor and von Willebrand factor. Integr Biol (Camb) 8: 813-820.
- Chen J, Verni CC, Jouppila A, et al. (2018) Dual antiplatelet and anticoagulant (APAC) heparin proteoglycan mimetic with shear-dependent effects on platelet-collagen binding and thrombin generation. Thromb Res 169: 143-151.
- Schneider SW, Nuschele S, Wixforth A, et al. (2007) Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci 8: 7899-7903.
- Liu G, Fang Y, Wu J (2013) A mechanism for localized dynamics-driven affinity regulation of the binding of von Willebrand factor to platelet glycoprotein Iba. J Biol Chem 13: 26658-26667.
- Diamond SL, Purvis J, Chatterjee M, et al. (2013) Systems biology of platelet-vessel wall interactions. Front Physiol 15: 4.
- Pujadas Mestres L, Lopez Vilchez I, Arellano Rodrigo E, et al. (2017) Differential inhibitory action of apixaban on platelet and fibrin components of forming thrombi: Studies with circulating blood and in a platelet-based model of thrombin generation. PLoS One 12: e0171486.
- Yin W, Bond K, Rouf F, et al. (2015) Altered flow changes thrombin generation rate of circulating platelets. Ann Biomed Eng 43: 2827-2837.
- Meza D, Shanmugavelayudam SK, Mendoza A, et al. (2017) Platelets modulate endothelial cell response to dynamic shear stress through PECAM-1. Thromb Res 150: 44-50.
- Meza D, Musmacker B, Steadman E, et al. (2019) Endothelial cell biomechanical responses are dependent on both fluid shear stress and tensile strain. Cell Mol Bioeng 12: 311-325.
- Kirchhofer D, Sakariassen KS, Clozel M, et al. (1993) Relationship between tissue factor expression and deposition of fibrin, platelets, and leukocytes on cultured endothelial cells under venous blood flow conditions. Blood 15: 2050-2058.
- Yau JW, Teoh H, Verma S (2015) Endothelial cell control of thrombosis. BMC Cardiovasc Disord 19: 130.
- Siegel-Axel DI, Gawaz M (2007) Platelets and endothelial cells. Semin Thromb Hemost 33: 128-135.
- Geenen ILA, Molin DGM, van den Akker NMS, et al. (2015) Endothelial cells (ECs) for vascular tissue engineering: Venous ECs are less thrombogenic than arterial ECs. J Tissue Eng Regen Med 9: 564-576.
- Rubenstein DA, Yin W (2009) Glycated albumin modulates platelet susceptibility to flow induced activation and aggregation. Platelets 20: 206-215.
- Rubenstein DA, Yin W (2010) Quantifying the effects of shear stress and shear exposure duration regulation on flow induced platelet activation and aggregation. J Thromb Thrombolysis 30: 36-45.
- Yin W, Rubenstein DA (2009) Dose effect of shear stress on platelet complement activation in a cone and plate shearing device. Cell Mol Bioeng 2: 274-280.
- Yin W, Rouf F, Shanmugavelayudam SK, et al. (2014) Endothelial cells modulate platelet response to dynamic shear stress. Cardiovasc Eng Technol 5:145-153.
- Ruggeri ZM (1994) Platelet adhesion under flow. Microcirculation 16: 58-83.
- Hansen CE, Qiu Y, Mc Carty OJT, et al. (2018) Platelet Mechanotransduction. Annu Rev Biomed Eng 20: 253-275.
- Rana A, Westein E, Niego B, et al. (2020) Shear-Dependent platelet aggregation: Mechanisms and therapeutic opportunities. Front Cardiovasc Med 6: 141.
- Galbusera M, Zoja C, Donadelli R, et al. (1997)Fluid shear stress modulates von Willebrand factor release from human vascular endothelium. Blood 90: 1558-1564.
- Rusu L, Minshall RD (2018) Endothelial cell Von Willebrand factor secretion in health and cardiovascular disease. Endothelial dysfunction - old concepts and new challenges. Intech Open.
- Savage B, Saldívar E, Ruggeri ZM (1996) Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84: 289-297.
- Savage B, Almus-Jacobs F, Ruggeri ZM (1998) Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell 94: 657-666.
- Doggett TA, Girdhar G, Lawshé A, et al. (2002) Selectin-like kinetics and biomechanics promote rapid platelet adhesion in flow: The GPIb(alpha)-vWF tether bond. Biophys J 83: 194-205.
- Jesty J, Yin W, Perrotta P, et al. (2003) Platelet activation in a circulating flow loop: Combined effects of shear stress and exposure time. Platelets 14:143-149.
- Xu XR, Carrim N, Neves MAD, et al. (2016) Platelets and platelet adhesion molecules: Novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb J 14: 29.
Corresponding Author
Wei Yin, PhD, Bioengineering Building, Room 109, Stony Brook University, Stony Brook, NY 11794, USA, Tel: 631-632-1897.
Copyright
© 2022 Steadman E. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.