Utility of Exome Sequencing Databases in Validating Genetic Variants Associated with Multiple Endocrine Neoplasia
Abstract
Background
Multiple Endocrine Neoplasia (MEN) syndromes and Familial Medullary Thyroid Cancer have a well-documented genetic origin; However, it is not always clear whether genetic variation represents a pathologic vs. normal rare variant.
Objectives
We aim to assess the validity of published variants using online exome sequencing databases, and to identify undiscovered variants which potentially cause disease.
Methods
A literature search of PubMed, OMIM, and online MEN2 databases was conducted to include genetic variants thought to be causative of MEN. Published variants were compared against the Exome Variant Server (EVS) and 1000 Genomes Project.
Results
There were 40 publications which yielded 170 unique variants implicated in the pathogenesis of MEN. Of these, 47 variants were found within exome sequencing databases. Six published variants were found within sequenced populations at inappropriately high frequencies. Exome sequencing data analysis revealed seven potentially causative variants not found in the literature.
Conclusions
The current MEN literature is robust in that most published variants are absent or rare within exome sequencing databases. However, some variants may be inappropriately implicated with causing disease. Additionally, the EVS identified undescribed variants which may be of interest.
Keywords
Exome sequencing, Exome variant server, 1000 Genomes project, Medullary thyroid cancer
Introduction
Multiple Endocrine Neoplasia (MEN) syndromes and Medullary Thyroid Cancer (MTC) have a pathogenesis with a well-documented genetic origin. This group of diseases is divided in to MEN1, MEN2a, and MEN2b based on the specific constellation of tumors and phenotype that arises. Mutations in the MENIN and RET genes cause MEN1 and MEN2a/b, respectively, and specific mutations significant prognostic implications. As attention has turned toward understanding the genetics of tumorigenesis, a library of implicated mutations has been generated. Most of these take the form of Single Nucleotide Polymorphisms (SNPs) that result in missense mutations. While some of these mutations have a very strong link with development of the phenotype, many published variants occur too infrequently within study populations to reliably determine a causative role in disease. The process by which these mutations are deemed abnormal has traditionally relied on inference stemming from the degree of sequence conservation across species [1,2]. This method of determining genetic normality can unintentionally classify non-pathologic variants as potentially disease-causing, while individuals may harbor rare variants of unknown functional consequence [3-5]. Therefore, it will become increasingly important to classify variants accurately for prognostic and therapeutic decision-making.
Recently, advancements in genomic sequencing have made it possible to assess genetic variation across broad populations. It has become clear that individuals may harbor rare variants of unknown functional consequence. To reduce this problem, it has been proposed that genome databases such as the 1000 Genomes Project or the Exome Variant Server may be used as controls when investigating genetic variation in individuals with rare disease states [3,6-8]. This has previously been demonstrated with Mendelian diseases [9]. These databases are compiled from high-throughput sequencing data from large populations and mirror the genetic variability present in the general population. Charapenova, et al. examined the state the literature of genetic pediatric epilepsy syndromes using these databases in order to make a statement on the robustness of prior publications [3]. As with MEN, analysis of small sample sizes may incorrectly classify mutations as pathogenic when they in fact represent a rare normal human variant. As these technologies and genetic databases develop, the information can be utilized to minimize reporting errors associated with disease pathogenesis. In addition to compiling allelic frequencies of sequenced variants, certain databases utilize the protein folding software PolyPhen to predict the impact of amino acid substitutions on protein structure and function for each sequenced variant. These predictions are generated based on computer modeling of protein folding and the subsequent anticipated impacts on domains vital to protein function. The combined use of PolyPhen scores and variant allele frequency may allow for better characterization of novel variants. In the present study, we aimed to validate the use of exome sequencing databases for the broad and rapid evaluation of novel mutations by applying this method to previously published MEN-causing gene variants.
Materials and Methods
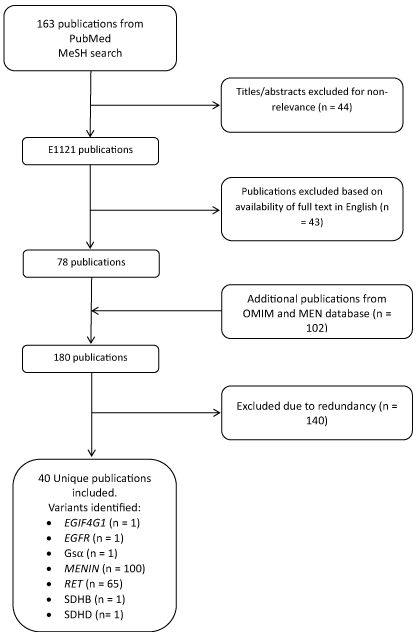
A systematic review of English literature conducted with the MeSH keywords ''Multiple Endocrine Neoplasia" AND "Point Mutation" yielded 163 publications. The references of these papers were reviewed for key articles. Additionally, the OMIM database entries for MEN1, MEN2, and sporadic or familial Medullary Thyroid Cancer (sMTC, FMTC) were reviewed for additional references. We also included the published MEN2 Database variants within the analysis [10]. Variants were tabulated and queried in the Exome Variant Server and 1000 Genomes Project databases. The allelic frequency and PolyPhen scores of the obtained variants were then recorded. A Minor Allele Frequency (MAF) of 0.005 was used as the threshold above which the variant is considered too common to be directly responsible for pathogenesis given the known disease prevalence [4,5].
Each gene implicated by the literature search was also analyzed directly within the EVS. The total number of variants found within the sample population was tabulated. Those variants with PolyPhen scores of "Possibly Damaging" or "Probably Damaging" were tabulated and included as well as those with a MAF > 0.005.
Results
Literature review and presence of identified variants within exome sequencing databases
The literature review produced 40 publications which met inclusion criteria (Figure 1). There were 170 unique variants across 7 different genes reported to be involved in the pathogenesis of MEN syndromes (Table 1). Of these, 100 variants were located within MENIN (previously called MEN1) associated with MEN1 syndrome. Only a single published variant, R171W, appeared in exome sequencing data and was predicted to be "Possibly Damaging" according to PolyPhen. It was found at an expected low allelic frequency of 0.000308. Additionally, 65 published variants were found within the RET gene associated with MEN2A, MEN2B, sMTC, or FMTC. Of those published variants, 46 were found within the exome sequencing databases (Table 2) with only 6 variants previously reported as "Damaging" found at MAF > 0.005 (Table 3). Of these 6 variants, five were synonymous mutations, and all have been implicated in sMTC pathogenesis. The variant G691S was associated with a MAF of 0.157 and is predicted to be benign [11]. Of the 38 published variants with expectedly low frequencies, 27 were predicted to damaging to the final gene product by PolyPhen, resulting in 33 total damaging variants of any MAF identified within the databases.
Assessment of overall variability of target genes within exome sequencing databases
The overall variance in each implicated gene was summarized (Table 4). EIF4G1 demonstrated the highest variability, followed by EGFR, with 339 and 317 variants respectively. Each gene had a total of 55 variants predicted to be damaging. Within EIF4G1, the variant R1223H was associated with a MAF of 0.0113 and predicted to be damaging by PolyPhen. RET had a relatively intermediate level of variability with 244 total variants. Of these, 47 were predicted to be damaging. There were three variants predicted to be damaging which also appeared at a MAF > 0.005. MENIN had a relatively low level of variance with only 72 variants within the database populations, 15 of which were predicted to be damaging. Only a single variant, R176Q, was both predicted to be damaging and appeared at a high MAF. The Gsp oncogene is a variant of GNAS. This gene is associated with 270 variants, only 11 of which are predicted to be damaging and none of which appear at high MAF. SDHB and SDHD have been implicated in MEN2B and were associated with 62 and 20 variants each. Of these, 6 and 5 are predicted to be damaging. Each gene had 1 variant appearing at a high MAF. None of these variants appeared among those found in our literature search (Table 5).
Discussion
Ideally, all variants under consideration must be germline rather than acquired and accumulated within the tumor cells. Much of the available literature on cancer genetics includes tumor genetics and acquired mutations. These are not necessarily amenable to analysis by population genetics among otherwise healthy persons. However, an important caveat to this statement is that rare somatic mutations may play a questionable role in a disease process if they are found at high rates among healthy populations. The MEN family of diseases was selected initially because of the relatively high penetrance of disease resulting in a more robust body of published variants that fit this requirement. Nevertheless, we believe that utilization of exome sequencing databases will play an increasing role in the validation of newly discovered germline mutations implicated in the pathogenesis of any hereditary cancer.
The present article represents a non-exhaustive systematic review of the published literature on the genetic basis of Multiple Endocrine Neoplasias. As previously stated, the goal was to critically evaluate the current state of the literature utilizing open-access databases such as EVS. The current gold standard of assessing abnormality of novel genetic variance relies on conservation across species. We aimed to adapt a method previously described in the pediatric neurology literature to diseases relevant to oncologic surgery [3]. Most the published variants collected here were absent from the EVS. Of those that appeared, the majority were present at sufficiently low rates such that they could represent undiagnosed individuals. Because EVS was initially designed with a focus on cardiopulmonary and hematologic disorders, this population may harbor individuals with undiagnosed MEN diseases. It is assumed that the frequency with which these individuals appear in such databases should mirror that of the general population, however this cannot be guaranteed [7,8,12]. The low rate at which published variants do appear highlights the robustness of the current literature. As expected, many key mutations previously discussed in the literature did not appear within the exome sequencing databases, most notably are those with prognostic and treatment implications. Specific RET codons include 883, 918, and 922 that drive recommendations for the most aggressive treatment including thyroidectomy before 6 months of age. The absence of such mutations in these populations is encouraging considering the potential unnecessary morbidity that could be associated with falsely attributing prognostic value to them. However, those variants which did appear in the databases at unexpectedly high rates raise important questions about the genetic pathogenesis of MEN syndromes and endocrine cancers. We included in this study a summary of SNPs which have been previously implicated in sMTC which yield silent mutations. For example, the variant G691S was associated with a MAF of 0.157 and is predicted to by PolyPhen to be benign, although in one study it was more prevalent in patients with sMTC versus healthy controls [11]. The authors who previously published these variants note that there is potential that the DNA-level mutation may impact splice variants which act to enhance or silence expression [13]. However, many of these variants occur at a greater allelic frequency than our 0.005 MAF threshold for variants associated with disease pathogenesis [11]. Therefore, either it is unlikely that these variants are involved in disease pathogenesis or they are subject to other epigenetic or regulatory effects that warrant further investigation. While this may reduce their prognostic significance, it is possible that they represent benign variants which co-segregate with other yet undescribed deleterious mutations.
A portion of our analysis focused on assessing the overall level of variation within an implicated gene rather than looking at specific variants themselves, which relied heavily on PolyPhen predictions. While there was a great deal of concordance among predictions in terms of rarity and severity of the deleterious effect, a subset of variants within the database were associated with a high PolyPhen score for the given MAF. None of these variants appeared at extraordinarily high MAF, and individuals harboring these variants could represent those with undiagnosed disease. These variants may be targets of future investigation. However, it should be noted that PolyPhen scores may require further validation before being used solely to guide future research. Interestingly, a search for the sole MENIN variant yields a single publication regarding adrenal cortical cancer, which is not traditionally associated with MEN1 [14]. Of the three variants found within RET, R982C was mentioned once in a patient with MEN1 with MEN2-like features or rarely in combination with other variants [15].
Conclusions
The present article stands as a proof of concept for the use of exome sequencing databases to evaluate published variants implicated in the pathogenesis of diseases relevant to surgical oncology. Through a retrospective evaluation of the literature, we identified some well-published variants present in the sequenced populations at unexpectedly high rates. The validity of such variants with regards to their role in disease is called into question. This illustrates how these databases allow for rapid evaluation of novel mutations and an opportunity for investigators to quickly validate their findings. An independent analysis of EVS data revealed several mutations which have not yet been implicated in associated MEN syndromes. Further work is warranted to determine what role such variants play in the disease process. These findings may lead to novel genetic targets for further research.
Grant
None.
Disclosures/Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- Bejerano G, Pheasant M, Makunin I, et al. (2004) Ultraconserved elements in the human genome. Science 304: 1321-1325.
- (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562.
- Cherepanova NS, Leslie E, Ferguson PJ, et al. (2013) Presence of epilepsy-associated variants in large exome databases. J Neurogenet 27: 1-4.
- Nelson MR, Wegmann D, Ehm MG, et al. (2012) An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337: 100-104.
- Tennessen JA, Bigham AW, O'Connor TD, et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64-69.
- Piton A, Redin C, Mandel JL (2013) XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet 93: 368-383.
- 1000 Genomes Project Consortium, Auton A, Brooks LD, et al. (2015) A global reference for human genetic variation. Nature 526: 68-74.
- Exome Variant Server.
- Maria Delio, Kunjan Patel, Alex Maslov, et al. (2015) Development of a targeted multi-disorder high-throughput sequencing assay for the effective identification of disease-causing variants. PLoS One 10: e0133742.
- Margraf RL, Crockett DK, Krautscheid PM, et al. (2009) Multiple endocrine neoplasia type 2 RET protooncogene database: Repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat 30: 548-556.
- Elisei R, Cosci B, Romei C, et al. (2004) RET exon 11 (G691S) polymorphism is significantly more frequent in sporadic medullary thyroid carcinoma than in the general population. J Clin Endocrinol Metab 89: 3579-3584.
- Auer PL, Reiner AP, Wang G, et al. (2016) Guidelines for large-scale sequence-based complex trait association studies: Lessons learned from the nhlbi exome sequencing project. Am J Hum Genet 99: 791-801.
- Figlioli G, Landi S, Romei C, et al. (2013) Medullary Thyroid Carcinoma (MTC) and RET proto-oncogene: Mutation spectrum in the familial cases and a meta-analysis of studies on the sporadic form. Mutat Res 752: 36-44.
- Schulte KM, Mengel M, Heinze M, et al. (2000) Complete sequencing and messenger ribonucleic acid expression analysis of the MEN I gene in adrenal cancer. J Clin Endocrinol Metab 85: 441-448.
- Diala El-Maouche, James Welch, Sunita K Agarwal, et al. (2016) A patient with MEN1 typical features and MEN2-like features. Int J Endocr Oncol 3: 89-95.
- Shimazu S, Nagamura Y, Yaguchi H, et al. (2011) Correlation of mutant menin stability with clinical expression of multiple endocrine neoplasia type 1 and its incomplete forms. Cancer Sci 102: 2097-2102.
- Kameyama K, Okinaga H, Takami H (2004) RET oncogene mutations in 75 cases of familial medullary thyroid carcinoma in Japan. Biomed Pharmacother 58: 345-347.
- Komminoth P, Muletta-Feurer S, Soltermann A, et al. (1996) Detection of RET-proto-oncogene mutations in the diagnosis of Type 2 endocrine neoplasia (MEN 2). Schweiz Med Wochenschr 126: 1329-1338.
- Cirafici AM, Salvatore G, De Vita G, et al. (1997) Only the substitution of methionine 918 with a threonine and not with other residues activates RET transforming potential. Endocrinology 138: 1450-1455.
- Krampitz GW, Norton JA (2014) RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer 120: 1920-1931.
- Benazzouz B, Hafidi A, Benkhira S, et al. (2008) C634R mutation of the protooncongene RET and molecular diagnosis in multiple endocrine neoplasia type 2 in a large Moroccan family. Bull Cancer 95: 457-463.
- Dvorakova S, Vaclavikova E, Duskova J, et al. (2005) Exon 5 of the RET proto-oncogene: A newly detected risk exon for familial medullary thyroid carcinoma, a novel germ-line mutation Gly321Arg. J Endocrinol Invest 28: 905-909.
- Kasprzak L, Nolet S, Gaboury L, et al. (2001) Familial medullary thyroid carcinoma and prominent corneal nerves associated with the germline V804M and V778I mutations on the same allele of RET. J Med Genet 38: 784-787.
- Cranston A, Carniti C, Martin S, et al. (2006) A novel activating mutation in the RET tyrosine kinase domain mediates neoplastic transformation. Mol Endocrinol 20: 1633-1643.
- D'Aloiso L, Carlomagno F, Bisceglia M, et al. (2006) Clinical case seminar: In vivo and in vitro characterization of a novel germline RET mutation associated with low-penetrant nonaggressive familial medullary thyroid carcinoma. J Clin Endocrinol Metab 91: 754-759.
- Frank-Raue K, Machens A, Scheuba C, et al. (2008) Difference in development of medullary thyroid carcinoma among carriers of RET mutations in codons 790 and 791. Clin Endocrinol 69: 259-263.
- Eng C, Clayton D, Schuffenecker I, et al. (1996) The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276: 1575-1579.
- Fazioli F, Piccinini G, Appolloni G, et al. (2008) A new germline point mutation in Ret exon 8 (cys515ser) in a family with medullary thyroid carcinoma. Thyroid 18: 775-782.
- Colombo-Benkmann M, Li Z, Riemann B, et al. (2008) Characterization of the RET protooncogene transmembrane domain mutation S649L associated with nonaggressive medullary thyroid carcinoma. Eur J Endocrinol 158: 811-816.
- Da Silva AM, Maciel RM, Da Silva MR, et al. (2003) A novel germ-line point mutation in RET exon 8 (Gly(533)Cys) in a large kindred with familial medullary thyroid carcinoma. J Clin Endocrinol Metab 88: 5438-5443.
- Shifrin AL, Xenachis C, Fay A, et al. (2009) One hundred and seven family members with the rearranged during transfection V804M proto-oncogene mutation presenting with simultaneous medullary and papillary thyroid carcinomas, rare primary hyperparathyroidism, and no pheochromocytomas: Is this a new syndrome--MEN 2C? Surgery 146: 998-1005.
- Pacini F, Romei C, Miccoli P, et al. (1995) Early treatment of hereditary medullary thyroid carcinoma after attribution of multiple endocrine neoplasia type 2 gene carrier status by screening for ret gene mutations. Surgery 118: 1031-1035.
- Gimm O, Marsh DJ, Andrew SD, et al. (1997) Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J Clin Endocrinol Metab 82: 3902-3904.
- Fink M, Weinhüsel A, Niederle B, et al. (1996) Distinction between sporadic and hereditary medullary thyroid carcinoma (MTC) by mutation analysis of the RET proto-oncogene. "Study Group Multiple Endocrine Neoplasia Austria (SMENA)". Int J Cancer 69: 312-316.
Corresponding Author
Tyler J Mouw, MD, Department of Surgery, The University of Kansas Medical Center, USA, Tel: 913-588-6284.
Copyright
© 2018 Mouw TJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.