Key Amino Acids in Understanding Evolutionary Characterization of Mn/Fe-Superoxide Dismutase: A Phylogenetic and Structural Analysis of Proteins from Corynebacterium and Hosts
Abstract
Superoxide dismutases (SODs) are enzymes widely observed in nature and commonly studied to understand the protection of reactive oxygen species (ROS). This work aims to understand the conservation of and correlation among the enzymes from pathogens and hosts, using coevolution analysis of amino acids in Mn/Fe-Superoxide Dismutase.
Five amino acid sets were found. Some information about the functions of these amino acids was collected from the literature, which may help to better understand what the correlation of these amino acids may imply for the protein. Two pockets on the protein structure near the active site were identified, which contain some residues observed in the sets of correlated residues. The phylogenetic analysis contributed to the understanding of evolutionary differences and similarities between both organisms. It was possible to observe patterns of specific amino acids for each kind of organism and, by contrast, some amino acids that were conserved for all organisms, which are hypothesized to impact crucial functions such as folding and other specific functions.
Keywords
Evolutionary biology, Coevolution of amino acids, Sequence analysis, Structural biology, Molecular phylogeny, Molecular modeling
Background
Superoxide dismutases (SODs) are enzymes that catalyze the dismutation of superoxide into oxygen and hydrogen peroxide and, due to this, are extremely important as antioxidant defense in most cells exposed to oxygen [1,2] . These enzymes can be characterized into three distinct families that are unrelated in primary sequence. However, they have converged in evolution to utilize a redox-active metal to partition O2- into hydrogen peroxide and oxygen. These families are classified according to the metal ion cofactor, which includes the family of SODs that use a Ni ion to mediate the chemistry. These families include: (i) Cu/Zn-SODs, that use copper for catalysis and also bind a structural zinc atom; (ii) a rarer family of nickel-containing SODs called NiSOD; and (iii) an extensive Mn/Fe-SOD family that uses ionic manganese or iron [3-8]. According to data in the literature, NiSOD occurs only in prokaryotes. Cu/ZnSOD is described in eukaryotes and some prokaryotes, with some specific mutations on the proteins that are associated with important health issues such as neurodegenerative diseases like amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig's disease) [8]. FeSOD and MnSOD show a strong evolutionary relationship that probably belongs to a common ancestral gene [9]. Phylogenetic analysis of MnSOD suggests that information is shared in many different organisms, mainly in the cytoplasm of many bacteria and the mitochondria of eukaryotes [9]. Both enzymes have shown strong evolutionary divergence from each other, such that the two metals cannot functionally replace each other in Mn/FeSODs from most species.
Some studies have carried out the structural determination of SOD to understand the divergences between both enzymes [10-13]. In the Mn/FeSOD structures, the molecular architecture is composed of a C-terminal α/β domain and N-terminal helices that compose the polypeptide chain. In bacteria, the N-terminal domain is shown in dimers formation. The MnSOD in humans has a different structure, forming tetramers [12,14].
Whatever the differences in structure, MnSOD and FeSOD family members are widespread in biology and are so similar that they have probably evolved from a common ancestor before the divergence of eubacteria, archaebacteria, and eukaryotes over 3 billion years ago [15-20]. In prokaryotes, FeSOD is found in both aerobic and anaerobic bacteria [21]. On the other hand, MnSOD is found in aerobic bacteria and its transcription is up regulated upon exposure to oxygen, suggesting that it represents an elaboration [22]. In Eukaryotes, plants and protists have long been known to have FeSODs in their chloroplasts and mitochondria, respectively. In eukaryotic cells, this enzyme is critically required in mitochondria due to the high ROS (Reactive Oxygen Species) production within this organelle [23]. However, animals seem to lack FeSOD. Its absence in animal species has led researchers to propose that the FeSOD gene originated in plastids before moving to the nuclear genome [24].
Some prokaryotic species from Corynebacterium can survive in a hostile environment, for example, inside a bacterial phagosome within immune system cells, such as macrophages, probably due to the production of superoxide dismutase. Recently, some studies showed this enzyme could protect bacterial cells against ROS produced by biochemical mechanisms. Also, there are indications that in some pathogens, including some species of Corynebacterium, Mn/Fe-SOD may have an additional function in infection and colonization of the host [25]. Particularly, the species Corynebacterium pseudotuberculosis (Cp), which is the etiological agent of caseous lymphadenitis, a disease affecting mainly sheep and goats, can survive in this environment [26,27]. Both enzymes from bacteria and host are homologous. Structurally, it is possible to verify the common amino acids that are functionally coupled, i.e. coevolving. In the future, structural analysis will allow for identification of critical portions of protein, which could be used as drug targets. Thereby, some important questions arise. Are the amino acids of these proteins likely coevolving? Are there some properties of these proteins that are evolutionarily convergent or divergent?
Here, we intend to conduct the coevolution analysis of amino acids from Mn/Fe-Superoxide Dismutase of Corynebacterium and its host, with the aim to understand the conservation of and correlation among the enzymes from these organisms and consequently understand the evolutionary stages of this protein. Thus, our goal is to find subsets of specific amino acids, in different organisms, allowing a better understanding of protein structure and function. However, determining which residues of a protein contribute to a specific function is a significant challenge. In addition, several works have attempted to identify correlated residues from multiple sequence alignments [28-31]. These evolutionary studies have been applied to a few essential proteins and have provided new information about protein-protein interactions, ligand-receptor bindings, and the 3D protein structure [31-34].
Material and Methods
The sequences from C. pseudotuberculosis - Uniprot: D9Q547_CORP1 and Bos taurus - Uniprot: SODM_BOVIN were used in this work. We used The B. taurus sequence because it has more than 90% identity and coverage when aligned with other mammalian sequences (human, buffalo, horse, sheep, goat), and also because of the large amount of work with this sequence.
Protein sequences and multiple sequence alignment (MSA)
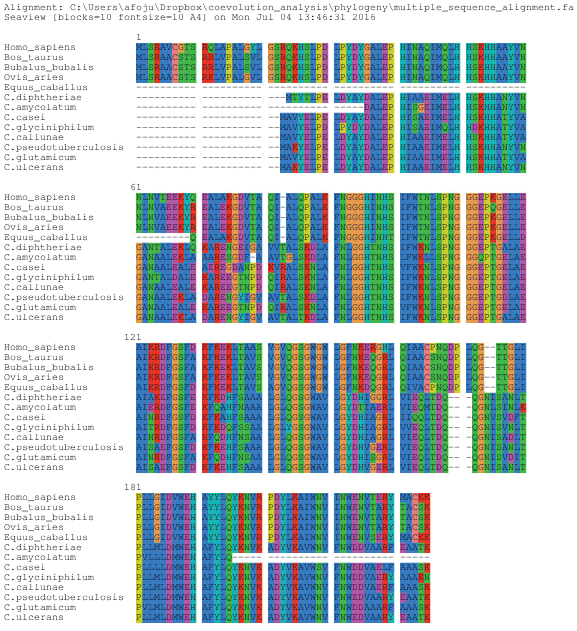
A multiple sequence alignment of the SOD protein family (Pfam code: PF00081) was conducted by (http://www.pfam.org/) from the Pfam database [35] and then subjected to three filtering procedures. First, to remove possible fragments, each sequence in the alignment was compared to a reference sequence. Sequences with fewer than 80% alignment coverage were removed from the alignment. Next, to remove poorly aligned sequences, those with less than 15% identity when compared to the reference sequence were also removed. Finally, in order to reduce phylogenetic bias, all sequences were compared to each other, and whenever two sequences shared higher than 90% identity, one of them was removed. The final alignment contained 225 sequences. The software package PFSTATS [36] was used for all procedures involving alignment filtering (pre-processing), conservation and correlation calculations (see below).
Statistical analysis (conservation and correlation)
The residue-position pairs were considered to be correlated if they passed the following thresholds: the correlation score [36] absolute value was higher than 10 (i.e., the p-value associated with the shift in frequency is lower than 10-10). The presence of one amino acid residue position (e.g., H33) must imply a resulting frequency for the other residue-position (e.g., L28) of at least 80% in case of correlation or at most 20% in the event of anti-correlation, and both positions must be present in at least 10% of the sequences as determined by the procedure described in [37]. A correlation network/graph can be built by defining nodes/vertices as residue-position pairs and connections/edges being present when the above thresholds are met. This network was decomposed by detecting connected components, resulting in five sets of correlated residues [36].
Phylogenetic analysis
The evolutionary model was determined using MEGA [38], which examines the alignment, obtains the closest evolutionary model and, thus, assists in building a tree. The popular WAG model, which combines the estimation of transition and scoring matrices by a maximum-likelihood approach, was selected as an evolutionary model. We used the tool Seaview [39], which is a multi-platform, graphical user interface for multiple sequence alignment and molecular phylogeny, in which the maximum likelihood method from PhyML software [40] and multiple sequence alignment from ClustalX was used [41]. Branch support consistencies were evaluated using the nonparametric bootstrap test [42] with 1000 replicates and the approximate likelihood ratio test (ALRT) [43]. Finally, the tree was edited highlighting the branches, organisms (sequences) and bootstrap value using Figtree [44,45].
Structure analysis
The comparative molecular modeling of proteins from C. pseudotuberculosis was performed with the software modeller [46]. Table 1 presents the details of the template structures that were selected by Blastp results, using information from NCBI [47] and PDB (Protein Data Bank) [48]. Structural images were produced using Pymol [49]. The Validations of models were done by the PROCHECK tool [50] that use the Ramachandran plot, the Discrete Optimized Protein Energy (DOPE) score [51], that is a statistical potential able to provide an energetic validation and finally by the RMSD obtained from a structural alignment with a protein with similar function, to provide an functional validation.
The tool DoGSiteScorer [52,53], which utilizes pattern recognition techniques for the identification of active sites, was used to find pockets and sub-pockets in the protein structure.
Results and Discussions
Residue conservation
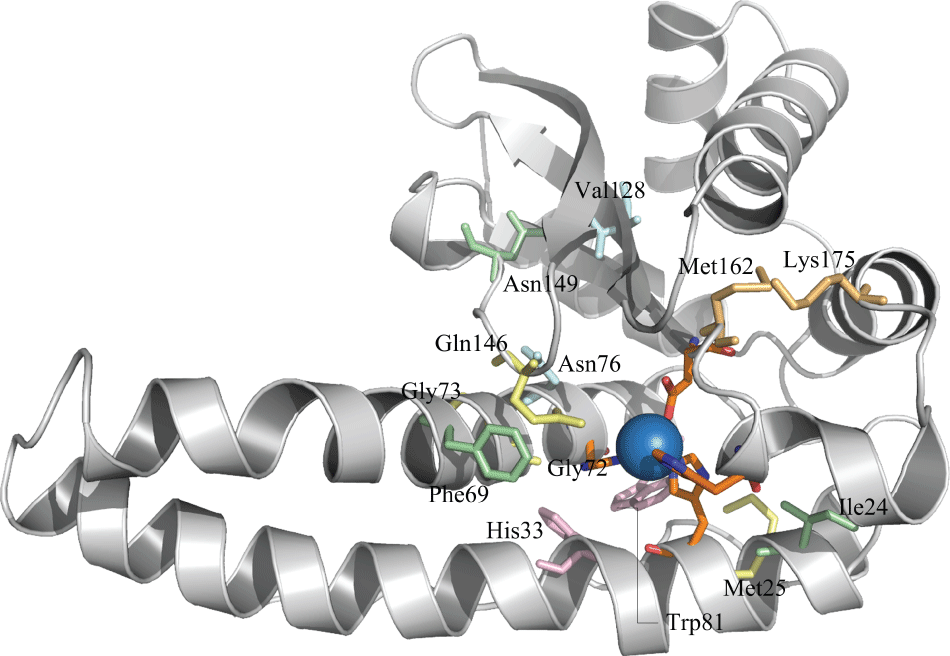
In order to investigate the possible relationship between statically coupled sequence positions, we applied conservation and correlation calculations to the protein family as a whole. Five amino acid sets were found, wherein seven residues were present in higher than 80% frequencies namely, Thr24, Asn69, Pro149, Gly72, Gly73, Met25, and Gln146 (numbers in superscript designate the position of the residues in the structure). The location of such residues in the tertiary structure of the representative superoxide dismutase can be seen in figure 1. The triad of histidines His28, His77 and His165 jointly with Asp161 form the active site.
Sets of correlated residue
Among the five amino acid sets, the first set contains three correlated amino acids, namely Tyr24, Asn69, and Pro149, where the number means the position in the structure. In our results, these three residues varied in the C. pseudotuberculosis protein sequence. Tyr24 increases the presence of Asn69 and Pro149 by more than 90%, in which its absence may result in the substitution of these amino acids. Our results show that Tyr24 is absent from 29.6% of all the sequences, which may help explain the substitutions at the other residues as well. In this study, the C. pseudotuberculosis sequence has the substitution Tyr24Ile, which could explain the other substitutions Asn69Phe and Pro149Asn in the same sequence. Curiously, the same event happens in the mammalian sequence as well.
The second set of correlated residues is comprised of four amino acids: Gly72, Gly73, Met25, and Gln146. The literature [54,55] discusses the importance of some residues involved in metal selectivity, which have been identified as Mn-specific. In our work, it was observed that all residues from the second set share the metal selectivity role. The importance of Gln146 was suggested in other studies, where Silverman's team [56] found that mutations affecting residues that hydrogen bond with the conserved active site Gln146 or His30 (in C. pseudotuberculosis structure His28) in the substrate access channel, result in very "longlived" inhibited complexes that accumulate to up to 20 times higher levels than WT Mn-SOD [57]. However, a mutant Mn-SOD presented a reduction in the formation of the inhibited complex, observing anti-proliferative properties and resulting in slower-growing tumors [58]. Additionally, other work [59] also noted that the residue Gln146 is a fundamental component of an extended hydrogen-bonding network around the active site. This group saw that a mutation of Gln146 to histidine or leucine results in the loss of more than 90% of the enzyme's activity. Furthermore, comparing the mutation of Gln146 and Tyr34, they observed that the Glutamine residue has a greater effect on protein structure than mutation of Tyr34, resulting in rearrangement of neighboring groups. For this reason, Gln146 has been implicated as playing an important role in the catalytic fine-tuning of these enzymes, largely based on its proximity to the metal center and its involvement in a conserved hydrogen-bonding pattern in the active site.
The third set of correlated residues contains the amino acids His33 and Trp81. Both residues are conserved in C. pseudotuberculosis and mammals. A study [13] analyzing the structure of Mn-SOD from Thermus thermophilus showed some information about this set of amino acids. It was observed that the residue His32 (His33 in C. pseudotuberculosis), together with others from this study, prevent access of solvent (and by analogy, substrate) to the metal or its ligands. Furthermore, Burley & Petsko [60] suggested that the residue Tyr36 can connected with Gln151, (Gln148 in the C. pseudotuberculosis sequence), to thus connect with the solvent, bound at Mn, which can provide a route by which the ionization of Tyr36 may interact with the charge on the metal-ligand. Studies from Burley and Petsko propose that the Trp87 residue (Trp81 in C. pseudotuberculosis) makes these contacts.
The fourth set of correlated residues is a compound of two amino acids: Asn76 and Trp128. However, Corynebacterium pseudotuberculosis has the variation Trp128Val. This replacement does not change the physicochemical characteristics and thus does not modify the possible connection between this residue and other amino acids. Some studies discuss the conservation of some specific amino acids, in which Trp128 is described as one of those present in either Mn-SOD or Fe-SO [61]. Xiang's works [62] introduced the possible function of Ans76 on protein structure. It was discussed that this amino acid can interact with the aromatic residue Phe136, to form an aromatic-polar internal chain interaction across the dimer interface situated not far from the entrance of the main substrate channel. However, our results do not corroborate this finding, in which the position 136 (122 in C. pseudotuberculosis) is not occupied by a Phenylalanine as seen [61], but by a Glutamine. Even without this substitution, the distance between Ans76 and Phe136 makes the molecular interaction between them impossible.
The fifth set contains two correlated amino acids, namely Val162 and Arg175. The pattern suggested in this set is observed in the mammalian Mn-SOD protein. These organisms have Arg175 followed by Val162, while in the Corynebacterium pseudotuberculosis Mn-SOD, there is Lys175 and Met162, as can be observed in the known multiple sequence alignment. In order to investigate these differences and the importance of each amino acid at these specific positions, some researchers search for effects of these residues on, for example, the structure and functional properties of superoxide dismutase as discussed next.
Gabbianelli, et al. by analyzing Propionibacterium shermanii's superoxide dismutase, observed that there is an effect of Lys175 mutation on the structure and functional properties of this protein, particularly in the structural stabilization of the active site [63]. Propionibacterium shermanii is very similar to our object of study, Corynebacterium pseudotuberculosis, belonging to the same phylum, Actinobacteria, as well as being gram-negative and able to establish a parasite-host relationship with mammals. It is known that there is a strict conservation of arginine at position 175, and both P. shermanii and C. pseudotuberculosis SODs are exceptions, wherein Arg175 is conservatively replaced by a lysine residue.
This high degree of conservation of arginine and lysine residues in prokaryotic and eukaryotic organisms, respectively, suggests that these residues play a major role in the enzyme-substrate interaction. It is known that Lys175 is involved not only in the electrostatic attraction of the substrate but also in the structural stabilization of the active site. Furthermore, inspection of the 3D structure suggests that Lys175 may play a relevant role in the substrate-enzyme interaction. Arg175 is also involved in catalytic differences when there is a substitution. Catalytic measurements show that the conservative substitution Lys175Arg produces only a small decrease in the activity and the structural rearrangements occurring at the level of the active site may be responsible for the further decrease in activity observed. Additionally, analysis of the X-ray structure of Mn/Fe-SODs [63] indicates that Lys175Arg is involved in several interactions stabilizing the structure of the active site channel [64-66].
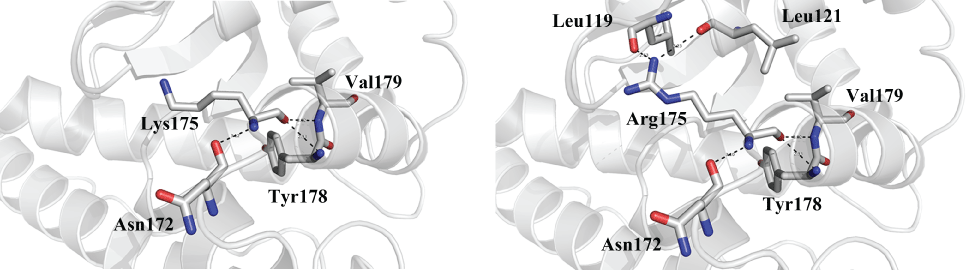
Noting these differences between the amino acids at position 175 in mammals and C. pseudotuberculosis, and that both are involved in practically the same processes, we hypothesize that the substitution that occurs at this position does not prevent any interaction between the enzyme and substrate for either the bacterial or mammalian cells. Indeed, it may even make it easier and more successful. Our analysis showed a few different interactions depend on the kind of amino acid at position 175 (Figure 2). It was observed that Lys175 could connect with Asn173, Tyr178, and Val179. Tyr178 and Val179 together form a part of one α-helix and Asn173 belong to a loop region. However, Arg175, in addition to having the same connections, can connect with two more residues, Leu119, and Leu121. Even though the variation Arg175Lys does not change the physiochemical characteristics of the amino acid, it modifies the number of connections depending on the type of amino acid. It is possible that this variation promotes a difference in protein structure by further strengthening the interaction among the amino acids of this region.
Another substitution described in the literature, which follows the same pattern as the Lys175Arg substitution, was also observed in the alignments, although it has not been highlighted by the graphs generated in our analysis. In the C. pseudotuberculosis MnSOD, each time we see a Lys175, we also see a Phe167, and in the mammalian superoxide dismutase protein, if there is an Arg175, there is also a Tyr167. This pattern is explained by the hydrophobic interactions involving Phe167 and the Lys175 aliphatic chain, which stabilizes the loop structure of the protein, forms part of the monomer-monomer interface and contributes one of the metal ligands (His165). In the case of the mammalian MnSODs, in which an arginine residue is present at position 175 and a tyrosine residue replaces Phe167, this substitution forms a hydrogen bond between its side chain and Arg175 [64].
These results agree with the statistical data that show how the presence of one amino acid can influence the presence of another. Our results indicate that 79.8% of all sequences have Val162, but this rate increases to 90.9% when Arg175 is present. In mammals, it was exactly the pattern observed, while in C. pseudotuberculosis, the position 162 is occupied by methionine.
It was observed that some amino acid sets might be in anticorrelation (Additional File 1). This allows us to interpret the probable difference between sets 2 and 4, which were seen in anti correlation with some other sets.
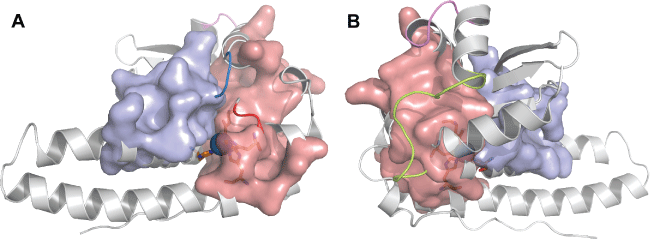
Pocket Analysis
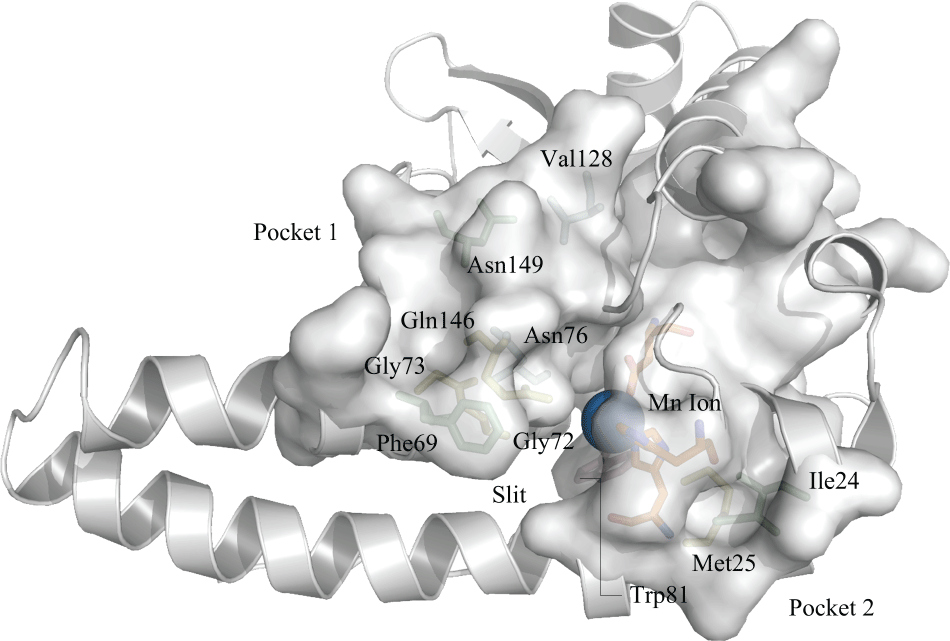
Two pockets were identified on the protein structure near the active site (Figure 3), which contain some residues observed in the sets of correlated residues. Pocket 1 is composed of Phe63, Asn149 (both belonging to the first set of correlated residues), Gln146, Gly72 (belonging to the second set), Asn76 and Val128 (belonging to the fourth set of correlated residues). Pocket 2 is composed of Ile24 (from the first set), Met25 (from the second set) and Trp81 (belonging to the third set). However, pocket 2 also contains the amino acids that make up the active site region. By analyzing the pocket regions, it is possible to see that the combination of both pockets creates a slit, which possibly favors the connection of the active site with the substrate. Furthermore, it is possible to determine that these correlated amino acids contribute to the formation of these pockets. No amino acids from the fifth set were observed in pocket formation. As discussed here, two positions have shown a variation in Mn-SOD structure, namely N149T and V128W. Both variations present the same physicochemical characteristics and due to this, it was not possible to find differences between the number of interactions and consequently their molecular conformations. Moreover, it is possible that this region in protein structure, which gives a favorable conformation for connection to the substrate and other ligands, be evolutionary conserved, due to some amino acids that make up this region that were shown to be evolutionarily related.
Key residues and phylogeny
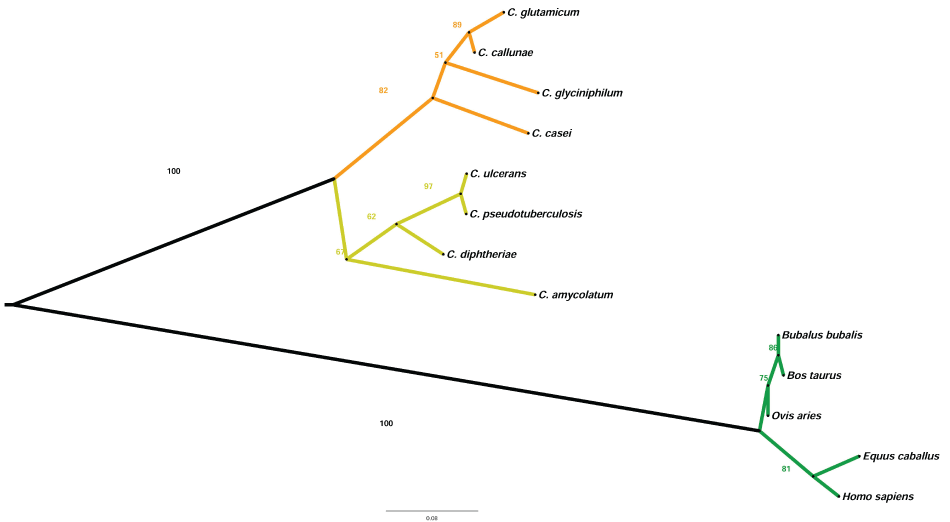
To understand the evolutionary difference present in our results concerning the coevolution of amino acids, the phylogenetic tree from the Mn-SOD protein sequences of some Corynebacterium species and their hosts was created and analyzed. The phylogenetic tree (Figure 4) shows the divergence between two high clades, bacteria and mammals. In the bacterial clade, the ramification of two subclades is clearly observed: one group of pathogenic bacteria, highlighted in yellow and another of non-pathogenic bacteria, highlighted in orange color. In the mammalian clade, the ramification of two subclades was also observed.
Analyzing the multiple alignments of Mn-SOD, it was possible to understand some divergences between bacteria and mammals (Figure 5). Our statistical data show some patterns of amino acids belonging to these different groups, which are N149T, V128W, M162V, and K175R. However, it was possible to identify, by MSA, thirty-four other patterns of amino acids that were not observed in our statistical analysis. They are: R33M, Q34A, H36Y, S37E, G46D, Q55E, A67N, N61G, L62A, E66L, L73R, Q86S, P87K, K90A, G93L, I0T, T104K, L119A, S140A, V143L, G149A, W150V, F163Y, N164D, N177D, T176S, V187M, Y192F, P201A, L204V, I207F, E213D and N214D. It is hence likely that the evolutionary division existing between these two organisms is due to the correlated amino acids substitutions that were found in our results and the observed patterns in the multiple sequence alignment. None of the correlated amino acids found in our statistical analysis results have a difference in physicochemical characteristics, although it was possible to observe differences in the number of interactions with other amino acids. However, analyzing the physicochemical characteristics of amino acids from MSA, it was noted that a vast majority of the amino acids substitutions changed the physicochemical characteristics. We hypothesize that these differences, analyzed by multiple sequence alignment, have an implication on the number of connections with other amino acids and probably on the three-dimensional structure of the protein as well.
It was observed that the pocket composition, which creates a catalytic slit in the enzyme, has some residues from patterns analyzed at the phylogenetic stage. They are L71G, T75I, K65P S115T, Q142A, and D145N. The substitutions Q142A and T75I change the physicochemical characteristics from polar to non-polar, consequently either reducing the number of connections with other neighboring amino acids or nullifying connection capability with neighboring amino acids. It is probable that such changes could destabilize some structural protein regions. Moreover, such analysis allowed for the identification of some patterns of amino acids that were fully conserved in all sequences. These amino acids were found in four loops (Figure 6) and constitute the pocket regions that were found in our results.
The first two (positions on structure in C. pseudotuberculosis 122-126 [144-149 in MSA] and 163-165 [188-190 in MSA]) are near the slit region of the active site, and others are "behind" (position on structure in C. pseudotuberculosis 84-96 [106-114 in MSA] and 104-107 [126-130]) the pocket. There is no information in the literature that describes the influence of these amino acids, so they remain poorly understood. Nevertheless, based on this analysis, we hypothesize that these loops may impart crucial/critical functions to the protein. It is likely that the loops near the active site either may be the key to the connection of substrate or have an influence on their stability, and that the loops found "behind" the pocket may be part of an allosteric site. Furthermore, an important observation about these amino acids is in their possible capacity for protein folding, in which we would suppose that the molecular geometry of this protein is due to the conservation of these loops.
Conclusion
In this study, we investigated the evolutionary similarities and differences between Superoxide Dismutase from Corynebacterium pseudotuberculosis and its host, considering the coevolution of some sets of amino acids and analysis of protein structure. It is known from the literature that the function of this protein is to protect the cell from reactive oxygen species (ROS), which can cause damage to many different biomolecules, like DNA. C. pseudotuberculosis and its host (mammalian cells) share a homologous protein, which complicates the study of this enzyme as a target to combat infection of this microorganism. However, in our results, it was possible to observe some sets of correlated amino acids sets that may influence the function of this protein, and that some amino acids of these sets varied in position depending on the organism. It was observed that the set of correlated amino acids has an impact on the formation of two pockets near the active site. In our analysis, it was possible to understand that these pockets create a slit that favors the connection of the substrate with these correlated amino acids and may affect the function of the protein. Phylogenetic analysis allowed for observation of how these sequences diverge across bacterial and mammalian clades, and it was possible to observe the pathogenic and non-pathogenic groups in the bacterial clade. Using the multiple sequence alignment, which was used to create the phylogenetic tree, it was possible to analyze some conserved patterns of amino acids that were not found to be statistically correlated, but were conserved in the alignment. Many of these correlated amino acids, depending on the organism, change the physicochemical characteristics, which may alter the number of interactions with neighboring amino acids and consequently stabilize specified regions. By contrast, it was possible to observe some converging protein patterns that were not lost during evolution. Four loop formations that are conserved in all sequences were identified, which may present some important function for the activity/regulation of the protein. We believe that such key amino acids may be exploited as good vaccine and drug target candidates, considering that some of these amino acids were observed from a specific organism. Our results show an opening for future research to assess the potential that these amino acids may have, for example, on the identification of epitopes in vaccine production and propose new studies using approaches in molecular docking, to identify chemical compounds that are able to interact with the region observed in this study. Furthermore, our study contributes to the understanding of the evolutionary steps of Manganese Superoxide Dismutase.
Funding
This work was supported by grants from Conselho nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes), Brazil.
Acknowledgements
The authors would like to acknowledge the institution UFMG and Jack Kames for the English review.
Competing Interests
The authors declare that they have no competing interests.
References
- Fridovich I (1997) Superoxide Anion Radical (O2), Superoxide Dismutases, and Related Matters. J Biol Chem 272: 18515-18517.
- McCord JM, Fridovich I (1969) Superoxide dismutase: An enzymic function for erythrocuprein. J Biol Chem 244: 6049-6055.
- Whittaker JW (2010) Metal uptake by manganese superoxide dismutase. Biochim Biophys Acta 1804: 298-307.
- Perry JJP, Hearn AS, Cabelli DE, et al. (2009) Human Manganese Superoxide Dismutase Tyrosine 34 Contribution to Structure and Catalysis. Biochemistry 48: 3417-3424.
- Rosen DR, Siddique T, Patterson D, et al. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59-62.
- Dupont CL, Neupane K, Shearer J, et al. (2008) Diversity, function and evolution of genes coding for putative Ni-containing superoxide dismutases. Environ Microbiol 10: 1831-1843.
- Wang Q, Johnson JL, Agar NYR, et al. (2008) Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol 6: e170.
- DiDonato M, Craig L, Huff ME, et al. (2003) ALS Mutants of Human Superoxide Dismutase Form Fibrous Aggregates Via Framework Destabilization. J Mol Biol 332: 601-615.
- MW Smith, RF Doolittle (1992) A Comparison of Evolutionary Rates of the Two Major Kinds of Superoxide Dismutase. J Mol Evol 34: 175-184.
- Parker MW, Blake CCF (1988) Crystal structure of manganese superoxide dismutase from Bacillus stearothermophilus at 2.4 Å resolution. J Mol Biol 199: 649-661.
- Carliozs A, Ludwigq L, Stallingsq C, et al. (1988) Iron superoxide dismutase. Nucleotide sequence of the gene from Escherichia coli K12 and correlations with crystal structures 263: 1555-1562.
- Borgstahl GE, Parge HE, Hickey MJ, et al (1992) The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell 71: 107-118.
- Ludwig ML, Metzger AL, Pattridge KA, et al. (1991) Manganese Superoxide Dismutase from Thermus thermophilus A Structural Model Refined at l-8 A Resolution. J Mol Biol 219: 335-358.
- Wagner UG, Pattridge KA, Ludwig ML, et al. (1993) Comparison of the crystal structures of genetically engineered human manganese superoxide dismutase and manganese superoxide dismutase from Thermus thermophilus differences in dimer-dimer interaction. Protein Sci 5: 814-825.
- Wolfe-Simon F, Grzebyk D, Schofield O, et al. (2005) The Role and Evolution of Superoxide Dismutases in Algae. J Phycol 41: 453-465.
- Pilon M, Ravet K, Tapken W (2011) The biogenesis and physiological function of chloroplast superoxide dismutases. Biochim Biophys Acta. Biochim Biophys Acta 1807: 989-998.
- Schmidt A, Gube M, Schmidt A, et al. (2009 ) In silico analysis of nickel containing superoxide dismutase evolution and regulation. J Basic Microbiol 49: 109-118.
- Ammendola S, Pasquali P, Pacello F, et al. (2008) Regulatory and structural differences in the Cu,Zn-superoxide dismutases of Salmonella enterica and their significance for virulence. J Biol Chem 283: 13688-13699.
- May BP, Dennis PP (1989) Evolution and Regulation of the Gene Encoding Superoxide Dismutase from the Archaebacterium Halobacterium cutirubrum. J Biol Chem 264: 12253-12258.
- Wintjens R, Noël C, May ACW, et al. (2004) Specificity and phenetic relationships of dismutases on the basis of structure and iron- and manganese-containing superoxide. J Biol Chem 279: 9248-9254.
- Santos WGDOS, Pacheco I, Liu M, et al. (2000) Purification and Characterization of an Iron Superoxide Dismutase and a Catalase from the Sulfate-Reducing Bacterium Desulfovibrio gigas. J Bacteriol 182: 796-804.
- Amo T, Atomi H, Imanaka T (2003) Biochemical Properties and Regulated Gene Expression of the Superoxide Dismutase from the Facultatively Aerobic Hyperthermophile Pyrobaculum calidifontis. J Bacteriol 185: 6340-6347.
- (1997) Outen BEVANH. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. 94: 514-519.
- Bowler C, Van Camp W, Van Montagu M, et al. (1994) Superoxide Dismutase in Plants. CRC Crit Rev Plant Sci 13: 199-218.
- Kehl-Fie TE, Chitayat S, Hood MI, et al. (2011) Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10: 158-164.
- Jung BY, Lee S-H, Kim H-Y, et al. (2015) Serology and clinical relevance of Corynebacterium pseudotuberculosis in native Korean goats (Capra hircus coreanae). Trop Animal Health Production 47: 657-661.
- Silva JW, Droppa-Almeida D, Borsuk S, et al. (2014) Corynebacterium pseudotuberculosis cp09 mutant and cp40 recombinant protein partially protect mice against caseous lymphadenitis. BMC Vet Res 10: 965.
- Gloor GB, Martin LC, Wahl LM, et al. (2005) Mutual information in protein multiple sequence alignments reveals two classes of coevolving positions. Biochemistry 44: 7156-7165.
- Mario A Fares, Simon A ATravers (2006) A novel method for detecting intramolecular coevolution: Adding a further dimension to selective constraints analyses. Genetics 173: 9-23.
- Dutheil J (2008) Detecting site-specific biochemical constraints through substitution mapping. J Mol Evol 67: 257-265.
- Yeang C, Haussler D (2007) Detecting Coevolution in and among Protein Domains. PLoS Comput Biol 3: e211.
- Shindyalov IN, Kolchanov NA, Sander C (1994) Can three-dimensional contacts in protein structures be predicted by analysis of correlated mutations? Protein Eng 7: 349-358.
- Wang ZO, Pollock DD (2007) Coevolutionary patterns in cytochrome c oxidase subunit I depend on structural and functional context. J Mol Evol 65: 485-495.
- Saraf MC, Moore GL, Maranas CD (2003) Using multiple sequence correlation analysis to characterize functionally important protein regions. Protein Eng 16: 397-406.
- Punta M, Coggill PC, Eberhardt RY, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40: D290-D301.
- Bleicher L, Lemke N, Garratt RC (2011) Using amino acid correlation and community detection algorithms to identify functional determinants in protein families. PLoS One 6: e27786.
- Dima RI, Thirumalai D (2006) Determination of network of residues that regulate allostery in protein families using sequence analysis. Protein Sci 15: 258-268.
- Tamura K, Stecher G, Peterson D, et al. (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30: 2725-2729.
- Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27: 221-224.
- Guindon S, Lethiec F, Duroux P, et al. (2005) PHYML Online - A web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33: W557-W559.
- Larkin MA, Blackshields G, Brown NP, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947-2948.
- Felsenstein J (1985) Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 39: 783-791.
- Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol 55: 539-552.
- Suchard MA, Rambaut A (2009) Many-Core Algorithms for Statistical Phylogenetics. Bioinformatics 25: 1370-1376.
- Pybus OG, Rambaut A, Harvey PH (2000) An integrated framework for the inference of viral population history from reconstructed genealogies. Genetics 155: 1429-1437.
- Eswar N, Webb B, Marti-Renom MA, et al. (2007) Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci.
- Benson DA, Karsch-Mizrachi I, Lipman DJ, et al. (2009) GenBank. Nucleic Acids Res 37: D26-D31.
- FC Bernstein, TF Koetzle, GJ Williams, et al. (1978) The Protein Data Bank: A Computer-based archival file for macromolecular structures. Arch Biochem Biophys 185: 584-591.
- Schrödinger LLC (2010) The PyMOL Molecular Graphics System, Version. 1.3r1.
- Morris AL, MacArthur MW, Hutchinson EG, et al. (1992) Stereochemical quality of protein structure coordinates. Proteins 12: 345-364.
- Melo F, Sánchez R, Sali A (2002) Statistical potentials for fold assessment. Protein Sci 11: 430-448.
- Volkamer A, Kuhn D, Rippmann F, et al. (2012) DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 28: 2074-2075.
- Volkamer A, Griewel A, Grombacher T (2010) Analyzing the topology of active sites: on the prediction of pockets and subpockets. J Chem Inf Model 50: 2041-2052.
- Kaprelyants AS, Gottschal JC, Kell DB (1993) Dormancy in non-sporulating bacteria. FEMS Microbiol Lett 10: 271-286.
- Keele BB Jr, McCord JM, Fridovich I (1970) Superoxide Dismutase from Escherichia coli B. A new manganese-containing enzyme. J Biol Chem 245: 6176-6181.
- Hearn AS, Fan L, Lepock JR, et al. (2004) Amino acid substitution at the dimeric interface of human manganese superoxide dismutase. J Biol Chem 279: 5861-5866.
- Hearn AS, Stroupe ME, Cabelli DE, et al. (2003) Catalytic and structural effects of amino acid substitution at histidine 30 in human manganese superoxide dismutase: Insertion of valine C gamma into the substrate access channel. Biochemistry 42: 2781-2789.
- Christopher AD, Amy S Hearn, Bradley Fletcher, et al. (2004) Potent antitumor effects of an active site mutant of human MnSOD: evolutionary conservation of product inhibition. J Biol Chem 279: 12769-12776.
- Edwards RA, Whittaker MM, Whittaker JW, et al. (2001) Outer sphere mutations perturb metal reactivity in manganese superoxide dismutase. Biochemistry 40: 15-27.
- Burley SK, Petsko GA (1988) Weakly polar interactions in proteins. Adv Protein Chem 39: 125-189.
- Osawa M, Yamakura F, Mihara M, et al. (2010) Conversion of the metal-speci fi c activity of Escherichia coli Mn-SOD by site-directed mutagenesis of Gly165Thr. BBA - Proteins Proteomics. Biochim Biophys Acta 1804: 1775-1779.
- Xiang H, Pan G, Vossbrinck CR, et al. (2010) A Tandem duplication of manganese superoxide dismutase in nosema bombycis and its evolutionary origins. J Mol Evol 71: 401-414.
- Gabbianelli R, Battistoni A, Polticelli F, et al. (1997) Effect of Lys175 mutation on structure function properties of Propionibacterium shermanii superoxide dismutase. Protein Eng 10: 1067-1070.
- Chan VWF, Bjerrum MJ, Borders CL (1990) Evidence that chemical modification of a positively charged residue at position 189 causes the loss of catalytic activity of iron-containing and manganese-containing superoxide dismutases. Arch Biochem Biophys 279: 195-201.
- Borders CL, Broadwater JA, Bekeny PA, et al. (1994) A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci 3: 541-548.
- Engler N, Ostermann A, Niimura N, et al. (2003) Hydrogen atoms in proteins: positions and dynamics. Proc Natl Acad Sci U S A 100: 10243-10248.
- Livingstone CD, Barton GJ (1993) Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation. Comput Appl Biosci 9: 745-756.
Corresponding Author
Alberto Oliveira, laboratório de Genética Celular e Molecular, Universidade Federal de Minas Gerais, 31270-901 - Belo Horizonte - MG, Brazil.
Copyright
© 2017 Oliveira A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.