Common Substrates of Medication-Resistant Temporal Lobe Epilepsy: Hippocampal Sclerosis, Focal Cortical Dysplasia, and Combined Pathologies
Abstract
Temporal lobe epilepsy (TLE) is an etiologically heterogeneous syndrome that constitutes the largest proportion of refractory partial epilepsies. The most common substrates of TLE include focal cortical dysplasia (FCD) in children and hippocampal sclerosis (HS) in adults, with significant overlap of lesion types across age groups. In contrast with the developmental nature of FCD pathology, HS is considered a post-natally acquired condition. This paper highlights the clinical and functional characteristics of each, with an introduction to their classification systems and relevant diagnostic information. While the underlying precipitants of either pathology are not precisely known, some proposed causative mechanisms are briefly reviewed, with additional discussion of combined FCD and HS vs. isolated lesions. Evidence for the functional relationship between co-occurring temporal FCD and HS is reviewed, including mesial temporal pathology as a secondary manifestation of seizure activity generated by the cortical lesion. It is postulated that childhood onset TLE associated with diagnosed HS is likely linked with neocortical dysplasia as the true underlying pathology that has escaped identification. Based on the epidemiology and diagnostic characteristics of these histological entities, it is hypothesized that the actual prevalence of focal cortical anomalies in the general population is underestimated while the rate at which FCD causes seizures is likely overestimated.
Keywords
Temporal Lobe Epilepsy, Focal Cortical Dysplasia, Hippocampal Sclerosis
Abbreviations
FCD: Focal Cortical Dysplasia; HS: Hippocampal Sclerosis; TLE: Temporal Lobe Epilepsy; MRI: Magnetic Resonance Imaging; CA: Cornu Ammonis
Introduction
Recurrent seizures affect more than 470,000 children and 3 million adults in the United States [1]. Of all patients with epilepsy, between 25 and 40 percent do not respond adequately to pharmacological management and are thus considered refractory [2,3]. While those with seizures originating from a focal area of the brain are at particular risk for pharmacoresistance [4] they often benefit from surgical intervention [5]. These benefits may include reduced seizure frequency or severity, improved cognitive and psychiatric outcomes, and improved overall quality of life [6].
Focal seizures are usually classified into syndromes based upon the lobe in which they originate, although they may be caused by a variety of brain pathologies. Temporal lobe epilepsy (TLE) is the most common form of epilepsy [7] and is etiologically heterogeneous. Adult temporal seizures most commonly arise from acquired damage to mesial temporal structures [8], while children may have either an underlying developmental lesion or postnatally acquired injury. Focal cortical dysplasia (FCD) is one of most commonly identified developmental malformations associated with intractable pediatric epilepsy [9,10], and is frequently associated with early seizure onset and neurological deficits [11]. A high percentage of children with developmental malformations of the temporal neocortex have additional pathology of the hippocampus [12,13].
Pathological Substrates
Hippocampal sclerosis
Also called mesial temporal sclerosis or Ammon's horn sclerosis, hippocampal sclerosis (HS) is the most commonly identified etiology of refractory seizures in adults [14]. Alterations to the normal hippocampal structures include neuronal loss with resultant atrophic changes, and gliotic scarring. In contrast to adults, HS in children with TLE is rarely found isolated from other pathologies [15]. In a pediatric surgical series of 136 TLE patients aged 3 months to 20 years, only 15% had HS [16]. The majority of HS seen in childhood is in older adolescents, rather than younger children [16,17], and although it does exist in younger children [13] the incidence in infants is rare [15].
While the exact etiology is unknown [18], there is a strong association between febrile convulsions in childhood and the later observation of HS [19]. Despite the lack of known causative mechanisms, animal models of damage to all hippocampal regions resulting from repeated kindled seizures suggest that HS is an acquired lesion [20]. HS further appears to be a progressive disorder, with greater severity of pathology associated with earlier onset and longer duration of seizures [21]. Gradual decreases in hippocampal volume have been noted in patients with continued seizures [22], likely related to uncontrolled inflammation caused by a cascade of molecular and cellular events associated with ongoing epileptiform discharges [23]. Wyler and colleagues [24] described a grading system for the progression of hippocampal sclerosis based on relative gliosis and neuronal cell loss:
• Grade I: Mild damage to CA1, CA3, and/or CA4, with less than 10% neuronal loss.
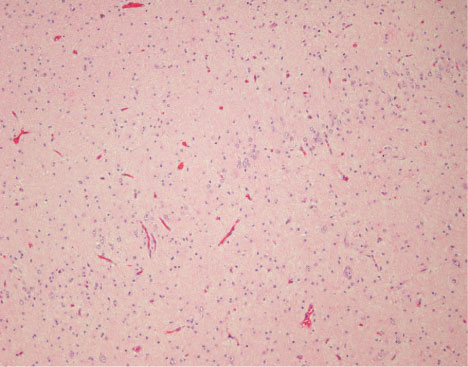
• Grade II: Moderate damage with 10 to 50% loss of neurons (Figure 1).
• Grade III: Severe damage with more than 50% neuronal dropout, sparing CA2.
• Grade IV: Marked mesial temporal damage, including greater than 50% cell loss involving all pyramidal cell layers. May also involve the dentate, subiculum, and parahippocampal gyrus.
More recently, an ILAE task force published a consensus report that further refined HS subtypes based on specific areas of cell loss and gliosis [25].
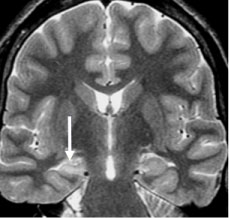
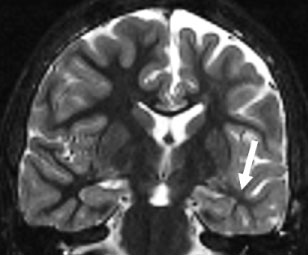
MRI evidence of HS includes volumetric loss, increased T2 signal, and loss of normal internal architecture [26] (Figure 2). While MRI visibility of HS often predicts seizure intractability [27], the severity of MR findings has not been directly correlated with pharmacological resistance [28]. Watson and colleagues [29] devised a volumetric MRI grading system for HS, yielding high correlation with Wyler's pathological grades. This grading method is based upon comparison between the measured volume of the epileptogenic hippocampus and that of the non-involved side. Although such volumetric measurement requires manually defining structural contours due to the complexity of the hippocampal formation, it allows for pre-operative estimation of HS severity.
Focal cortical dysplasia
FCD is a derangement of neuronal structures within the cortical tissue resulting from aberrant growth, migration, and/or proliferation of neural cells during the embryological stages of brain maturation [30]. Most noteworthy are architectural changes and disruption of columnar arrangement, with notable disruption of axonal alignment. Although showing some degree of correlation with adverse prenatal or perinatal events [31] FCD's appear to have less association with prenatal trauma history or genetic predisposition than other types of cortical malformation [32]. However, due to histological similarities between the balloon-cell FCD subtype and genetically-based syndromes such as Tuberous Sclerosis Complex, and notwithstanding differing clinical manifestations, a relationship between these pathologies has been suggested [33]. While data regarding the nature of dysplastic lesions continues to accumulate, much remains unknown about the mechanisms underlying FCD formation, with the possibility of multifactorial routes or multiple pathogenic processes that result in only a handful of terminal manifestations.
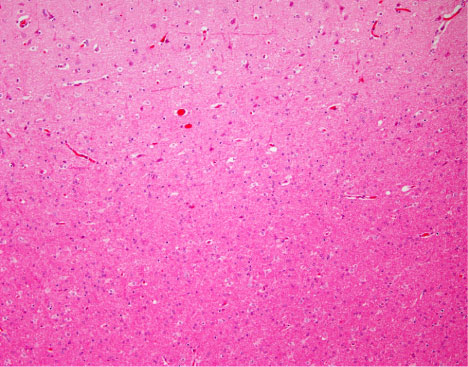
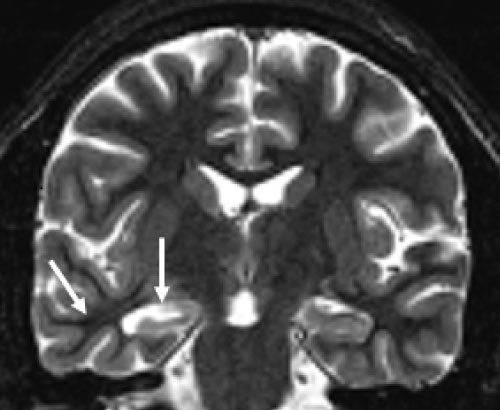
FCD pathology is identified by microscopic findings of disrupted laminar organization and abnormal neuronal morphology (Figure 3). While MRI findings typically include cortical thickening, blurring of the gray-white interface, and signal changes in the underlying white matter, FCD lesions vary in their visibility on MRI [34]. Although aberrant tissue is often visible on high-quality MR, many cases of FCD may remain undetectable by conventional imaging. Studies of cases with histologically proven FCD but no perceptibility on MRI suggest an occurrence rate ranging from 34% [35,36] to 42% [37]. Cortical dysplasia is generally best visualized on fluid-attenuated inversion recovery (FLAIR) sequences [38] but detection may be better with higher magnet field strength (e.g., 7-Tesla) [39].
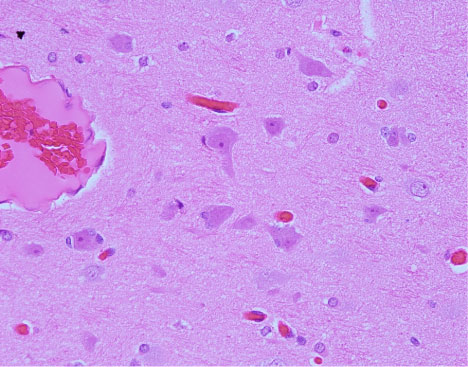
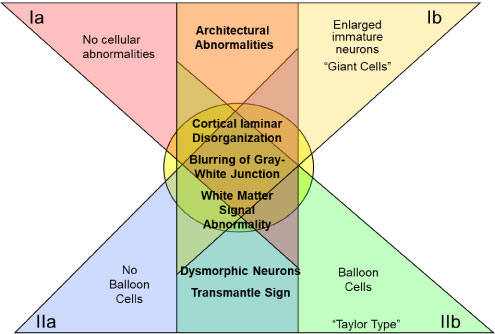
Taylor and Falconer recognized FCD as an underlying cause of seizures nearly half a century ago when they discovered disorganized cortical neurons and large, bizarre-shaped cells in ten epileptic patients [40]. Since then, considerable progress has been made toward understanding dysplastic changes to cortical tissue. Numerous studies have identified FCD as the most prevalent underlying cause of epilepsy within the temporal lobe [12] and other brain regions in children [41]. In 2004 Palmini, et al. documented the consensus of an expert panel based on specific variants of neocortical architecture and cellular histopathology [42]. Their proposed taxonomy comprised two major histological FCD divisions: Type I lesions include those with dyslamination of neocortical neurons with minor cellular abnormalities, while the major defining feature of type II is dysmorphic neurons or balloon cells. Each major type has two subdivisions (a and b). Type Ia FCD includes isolated architectural abnormalities of the cortical layers and Ib adds immature neurons (either "giant cell" or small diameter). Type II also includes architectural abnormalities, with IIa featuring the addition of dysmorphic neurons, and type IIb including both dysmorphic neurons and eosinophilic balloon cells [35,42] (Figure 4). Figure 5 graphically summarizes the Palmini classification system. More recently, Blumcke and colleagues [43] proposed a somewhat more expansive taxonomy that includes standalone FCD's as well as those occurring in tandem with other epileptogenic lesions (see combined pathology). They also added a third type I category, which contains abnormal lamination in both tangential and radial directions; however, unlike types Ia and Ib, type Ic does not occur in combination with any other pathologies. Barkovich's classification system [44,45] includes a wider range of developmental cortical malformations beyond FCD, such as lissencephaly, heterotopia, and schizencephaly [46].
Although the general mechanisms underlying the formation of FCD remain largely unknown, there is some evidence of links to cortical injury during the later prenatal stages of cortical maturation [46]. Differences between FCD subtypes are believed to result from varied ontological timing during neuronal migration or differentiation [44]. Some subtypes have also demonstrated regional associations within the cortex: Dysplasia within temporal areas is typically type I [47], while type IIB malformations with balloon cells (also known as "Taylor-type" cortical dysplasia) are most often in extratemporal regions, particularly the frontal lobes [36,48,49].
In general it is difficult to differentiate between FCD subtypes based solely on MRI features [50]; however, various imaging features have been identified across subtypes. At least one study has found better MRI imaging of Type II than Type I FCD [35] while others have shown equivalent visibility between subtypes [51]. Type IIb FCD may be more apparent on imaging than the other subtypes because balloon cells tend to generate greater signal change on MRI than dysmorphic or immature cells [52]. Krsek and colleagues [35] demonstrated that blurring of the junction between gray and white matter as well as the appearance of cortical thickening were much more evident on MRI in FCD type II, with lobar hypoplasia or atrophy more commonly observed in type I (Figure 6). In their surgical series, FCD type IIb lesions were more often restricted to a single lobe than were the other FCD subtypes [35].
FCD lesions are intrinsically Epileptogenic [53,54] and often lead to high seizure burden [30]. On the whole, dysplastic cortex is more conductive and hyperexcitable than normal brain tissue [12]. While dysplastic tissue embedded within integrated neural networks maintains some degree of functionality [56,57], neuronal tissue with abnormal cellular architecture is not as efficient as healthy brain tissue. Even at best, functional networks that include dysplastic areas underperform their healthy counterparts [58]. Blurring of the gray-white junction seen on MRI has been associated with cognitive morbidity [59], and the nature of cognitive alterations depend more upon lesion location and extent than involved FCD subtype [37]. While the electrophysiological properties of FCD's do vary based on their histopathology [53] (e.g., the balloon cells found in type IIb are electrically silent [60]), subtype does not appear to influence overall degree of epileptogenicity [35].
Combined pathology
As implied by the label, "dual pathology" denotes the presence of more than one type of defect. Blumcke type III cortical dysplasia includes FCD combined with various epileptogenic lesions, including HS (type IIIa), glioneuronal tumors (type IIIb), vascular malformations (type IIIc), and other lesions acquired in early life (type IIId) [43]. For purposes of the current paper, discussion has been limited to hippocampal sclerosis in tandem with FCD [61], consistent with type IIIa (Figure 7).
Combined HS and FCD pathologies are frequently detected in epilepsy surgery patients, but the incidence varies with population selection criteria. Overall studies of pediatric patients with TLE have revealed dual pathology in 58 to 79% of surgical patients [12,13]. Although temporal dual pathology in children typically consists of FCD and HS within the same temporal lobe [62] a large percentage of children and adolescents with histopathologically confirmed FCD have evidenced an additional pathology regardless of lobar location of dysplasia [35]. In pediatric cases with early seizure onset and confirmed FCD, 27% to 40% demonstrated co-occurring HS irrespective of lobar localization of seizure activity [36,62]; in a similar sample based on MRI evidence of FCD limited to the temporal lobes, this jumped to 87% [63]. Adults tend to evidence a lower incidence of combined pathology than children. In a surgical series of 33 adults with TLE, the majority had isolated HS, with only 48% evidencing dual pathologies [64]. However, MR spectroscopy in adults with known neocortical epileptic lesions (both temporal and extratemporal) has revealed subtle hippocampal abnormalities, even in the absence of gross hippocampal pathology detectable with MRI [65]. This evidence supports an etiological theory of dual pathology whereby damage to hippocampal structures may be secondary to repeated seizures originating from neocortical epileptic foci [63] although it would appear to be more applicable to pediatric than adult-onset TLE.
In a majority of patients with TLE, interictal focal slowing and EEG spikes correlate well with structural abnormalities detected by MRI [66]. In temporal lobe dual pathology, epileptic activity may be generated by the neocortical lesion, mesial temporal lesion, or both. In cases for which scalp EEG data does not sufficiently localize the seizure focus or is discordant with other findings, invasive EEG with placement of subdural or depth electrodes are often used and have revealed localized patterns of epileptogenic activity associated with dual pathologies. In twelve children and young adults with temporal lobe dual pathology, seizure activity originated from the mesial temporal area in 41% and from the temporal neocortex in 35%, while 22% demonstrated simultaneous discharges [67].
Epidemiological and Surgical Considerations
FCD is now known to be the cause of many epilepsies that were previously labeled as "cryptogenic" or having a suspected underlying basis that could not be identified. Although knowledge of cortical malformations has expanded greatly in the last two decades, the true prevalence of FCD's and incidence of epilepsy in those with such malformations remains unknown due to ascertainment bias and sampling errors. With the exception of isolated studies of normal volunteers, data is limited primarily to those who present with seizures or related issues. Since most individuals will never have cause for a brain MRI, much less a biopsy, the actual prevalence of focal cortical anomalies in the general population is likely underestimated. This idea is further supported by an increase in the reported prevalence of FCDs coinciding with improved imaging technology [68]. Likewise, ascertainment bias also leads to probable overestimation of the rate at which FCD's actually cause seizures; since our knowledge of individuals with FCD comes almost exclusively from an evaluation prompted by the presence of seizures, there may be many people with benign FCD producing no overt symptoms.
The high correlation between febrile convulsions in infancy and later findings of hippocampal sclerosis [69,70] also carries the implication of the febrile seizure as a precipitating event leading to subsequent HS development. In rat models with induced febrile convulsions, the presence of dysplastic cortex has been shown to increase the risk of later epilepsy by augmenting the effects of the febrile seizures upon hippocampal neurons [71]. Human data also suggests that early febrile convulsions in human infants may reflect pre-existing brain disease related to later evolution of partial seizures [72]. Along with very rapid seizure spread from neocortical to mesial temporal regions [73], this evidence is consistent with the relationship between HS and early febrile events in patients with temporal FCD [62,74], but without correlation to the degree of hippocampal atrophy [75]. Thus, the hippocampal component of temporal lobe dual pathology could be the consequence of seizure activity starting in hypersynchronous neocortex and propagating to the mesial temporal area, thereby inducing structural changes in the hippocampus that makes it more susceptible to epileptogenic activity [62,65].
Furthermore, because type I FCD predominates within the temporal lobe and tends to be less visible on MRI than type II, the neocortical component of a combined temporal pathology may escape detection in the absence of resection or biopsy to provide histopathological confirmation. Coupled with the high frequency of combined HS and FCD pathology (e.g., type IIIa) in children with intractable seizures, this raises the possibility that many, if not almost all, cases of pediatric TLE diagnosed as isolated HS may actually be a dual pathology in which the dysplastic neocortical component has not been identified. Despite evidence suggesting that HS arises secondary to cortical epileptogenic tissue, we cannot presently rule out a common underlying cause of lesions within both the neocortical and mesial temporal regions.
Surgical intervention has been shown superior to the prolonged pharmaceutical treatment of intractable temporal lobe epilepsy [76]. The ideal surgical resection strikes a balance between removing enough epileptic tissue to provide seizure control without harming eloquent cortex and creating a functional deficit, but the nature of dysplastic tissue makes this particularly challenging. In surgical series of FCD lesions, seizure freedom has varied from 50 to 72 percent of patients [77-80] regardless of FCD subtype. Although a number of surgical series have demonstrated greater overall reduction of seizure burden among individuals with type II pathology, [35,81-83] one study found better type I outcomes [84] with others showing little [85] or no difference [78] between type I and II FCD. In patients with dual pathology of the temporal lobe, resection of both the sclerotic hippocampus and neocortical lesion has resulted in greater seizure freedom than resection of the individual lesions [86-88]. Furthermore, dual pathology TLE outcomes are less favorable for cases in which the EEG and MRI data cannot be clearly interpreted [87].
While good surgical outcomes are associated with "complete resection" of the epileptogenic zone [31,41,58] the boundaries of dysplastic lesions can be particularly difficult to determine [89], especially with normal-appearing anatomical MR imaging. Even in "lesional" cases of FCD, the epileptogenic zone, conceptualized as the area of cortex that must be resected to produce seizure freedom [90], can be difficult to identify. Because dysplastic tissues have altered morphology or dispersion of individual neurons, and disorganization of neuronal layers [91], this zone is often larger than the area of dysplastic tissue typically visualized on MRI [52]. In cases of MR-negative FCD, delineating the epileptogenic zone may require invasive electrophysiological recording (i.e., electrocorticography and stereo EEG) as well as advanced neuroimaging techniques (i.e., SPECT, FDG-PET) to aid in the determination of appropriate resective boundaries [92,93].
Conclusions
Recent improvements in neuroimaging and histopathological techniques have given rise to a plethora of research focused on the pathologies underlying TLE; this explosion of knowledge has in turn facilitated improved surgical outcomes, yet numerous questions have yet to be resolved. While the precise etiology of HS remains unknown, it continues to be recognized as the most common pathology underlying adult-onset epilepsy and is generally accepted as a postnatally acquired condition. Conversely, histological characteristics of FCD evidence clear origins within the embryological period, although the precise causative factors remain undefined. Finally, emerging evidence suggests that co-occurring FCD and HS within the temporal lobe may be functionally interrelated rather than independent pathologies. Since milder forms of FCD may not be visible on magnetic resonance images ("MRI negative"), additional epidemiological studies of type IIIa dual pathology are likely benefit from histological ascertainment of neocortical tissues and consideration of specific hippocampal changes (e.g., volumetric and spectroscopic changes, as well as alterations to internal architecture).
Acknowledgement
A special thank you to Esperanza Pacheco-Jacome, M.D., Gabriel Chamyan, M.D., Carole Braithwaite, M.D., and Liset Pelaez, M.D. of Nicklaus Children's Hospital for their assistance with MRI and histopathological images.
References
- (2018) Epilepsy Fast Facts. Centers for Disease Control and Prevention.
- Kwan P, Arzimanoglou A, Berg AT, et al. (2010) Definition of drug resistant epilepsy: Consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia 51: 1069-1077.
- Snead OC (2001) Surgical treatment of medically refractory epilepsy in childhood. Brain Dev 23: 199-207.
- Donnadieu FR (2013) Pharmacoresistance and epilepsy. In: Rocha L, Cavalheiro EA, Pharmacoresistance in epilepsy: From genes and molecules to promising therapies Springer, USA, 1-9.
- Foldvary N, Bingaman W, Wyllie E (2001) Surgical treatment of epilepsy. Neurol Clin 19: 491-515.
- Spencer S, Huh L (2008) Outcomes of epilepsy surgery in adults and children. Lancet Neurol 7: 457-560.
- Devinsky O (2004) Diagnosis and treatment of temporal lobe epilepsy. Rev Neurol Dis 1: 2-9.
- Blumcke I (2009) Neuropathology of focal epilepsy: A critical review. Epilepsy Behav 15: 34-39.
- Harvey AS, Cross JH, Shinnar S, et al. (2008) Defining the spectrum of international practice in pediatric epilepsy surgery patients. Epilepsia 49: 146-155.
- Cepeda C, Andre VM, Levine MS, et al. (2006) Epileptogenesis in pediatric cortical dysplasia: The dysmature cerebral development hypothesis. Epilepsy Behav 9: 219-235.
- Chassoux F, Devaux B, Landre E, et al. (2000) Stereoelectroencephalography in focal cortical dysplasia: A 3D approach to delineating the dysplastic cortex. Brain 123: 1733-1751.
- Bocti C, Robitaille Y, Diadori P, et al. (2003) The pathological basis of temporal lobe epilepsy in childhood. Neurology 60: 191-195.
- Mohamed A, Wyllie E, Ruggieri P, et al. (2001) Temporal lobe epilepsy due to hippocampal sclerosis in pediatric candidates for epilepsy surgery. Neurology 56: 1643-1649.
- Lee YJ, Lee JS (2013) Temporal lobe epilepsy surgery in children versus adults: From etiologies to outcomes. Korean J Pediatr 56: 275-281.
- Mani J (2008) Temporal lobe epilepsy in children. J Pediatr Neurosci 3: 2-6.
- Wyllie E (1998) Surgical treatment of epilepsy in children. Pediatr Neurol 19: 179-188.
- Duchowny M, Levin B, Jayakar P, et al. (1992) Temporal lobectomy in early childhood. Epilepsia 33: 298-303.
- Thom M, Zhou J, Martinian L, et al. (2005) Quantitative post-mortem study of the hippocampus in chronic epilepsy: Seizures do not inevitably cause neuronal loss. Brain 128: 1344-1357.
- Cendes F, Andermann F, Dubeau F, et al. (1993) Early childhood prolonged febrile convulsions, atrophy and sclerosis of mesial structures, and temporal lobe epilepsy: An MRI volumetric study. Neurology 43: 1083-1087.
- Cavazos JE, Das I, Sutula TP (1994) Neuronal loss induced in limbic pathways by kindling: Evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci 14: 3106-3121.
- Fuerst D, Shah J, Kupsky WJ, et al. (2001) Volumetric MRI, pathological, and neuropsychological progression in hippocampal sclerosis. Neurology 57: 184-188.
- Fuerst D, Shah J, Shah A, et al. (2003) Hippocampal sclerosis is a progressive disorder: A longitudinal volumetric MRI study. Ann Neurol 53: 413-416.
- Yang T, Zhou D, Stefan H (2010) Why mesial temporal lobe epilepsy with hippocampal sclerosis is progressive: Uncontrolled inflammation drives disease progression? J Neurol Sci 29: 1-6.
- Wyler AR, Dohan FC, Schweitzer JB, et al. (1992) A grading system for mesial temporal pathology (hippocampal sclerosis) from anterior temporal lobectomy. J Epilepsy 5: 220-225.
- Blumcke I, Thom M, Aronica E, et al. (2013) International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A task force report from the ILAE Commission on diagnostic methods. Epilepsia 54: 1315-1329.
- Lewis DV (2005) Losing neurons: Selective vulnerability and mesial temporal sclerosis. Epilepsia 46: 39-44.
- Spooner CG, Berkovic SF, Mitchell LA, et al. (2006) New-onset temporal lobe epilepsy in children. Neurology 67: 2147-2153.
- Briellmann RS, Wellard RM, Masterson RA, et al. (2007) Hippocampal sclerosis: MR prediction of seizure intractability. Epilepsia 48: 315-323.
- Watson C, Nielsen SL, Cobb C, et al. (1996) Pathological grading system for hippocampal sclerosis: Correlation with magnetic resonance imaging-based volume measurements of the hippocampus. J Epilepsy 9: 56-64.
- Bast T, Ramantani G, Seitz A, et al. (2006) Focal cortical dysplasia: Prevalence, clinical presentation and epilepsy in children and adults. Acta Neurol Scand 113: 7-81.
- Krsek P, Jahodova A, Maton B, et al. (2010) Low-grade focal cortical dysplasia is associated with prenatal and perinatal brain injury. Epilepsia 51: 2440-2448.
- Montenegro MA, Guerierro MM, Lopes-Cendes I, et al. (2002) Interrelationship of genetics and prenatal injury in the genesis of malformations of cortical development. Arch Neurol 59: 1147-1153.
- Fassunke J, Blumcke I, Lahl R, et al. (2004) Analysis of chromosomal instability in focal cortical dysplasia of Taylor’s balloon cell type. Acta Neuropathol 108: 129-134.
- Hader W, Mackay M, Otsubo H, et al. (2004) Cortical dysplastic lesions in children with intractable epilepsy: Role of complete resection. J Neurosurg 100: 110-117.
- Krsek P, Maton B, Korman B, et al. (2008) Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol 63: 758-769.
- Tassi L, Colombo N, Garbelli R, et al. (2002) Focal cortical dysplasia: Neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 125: 1719-1732.
- Korman BM, Krsek P, Duchowny M, et al. (2013) Early seizure onset and dysplastic lesion extent independently disrupt cognitive networks. Neurology 81: 745-751.
- Tschampa HJ (2015) Magnetic resonance imaging of focal cortical dysplasia: Comparison of 3D and 2D fluid attenuated inversion recovery sequences at 3T. Epilepsy Res 116: 8-14.
- Colon AJ, vanOsch MJ, Buijs M, et al. (2016) Detection superiority of 7-T MRI protocol in patients with epilepsy and suspected focal cortical dysplasia. Acta Neurol Belg 116: 259-269.
- Taylor DC, Falconer MA, Bruton CJ, et al. (1971) Focal dysplasia of the cerebral cortex in epilepsy. J Neurol Neurosurg Psychiatry 34: 369-387.
- Krsek P, Maton B, Jayakar P, et al. (2009) Incomplete resection of focal cortical dysplasia is the main predictor of poor postsurgical outcome. Neurology 72: 217-223.
- Palmini A, Najm I, Avanzini G, et al. (2004) Terminology and classification of the cortical dysplasias. Neurology 62: S2-S8.
- Blumcke I, Thom M, Aronica E, et al. (2011) The clinicopathologic spectrum of focal cortical dysplasias: A concensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 52: 158-174.
- Barkovich AJ, Kuzniecky RI, Dobyns WB, et al. (1996) A classification scheme for malformations of cortical development. Neuropediatrics 27: 59-63.
- Barkovich AJ, Kuzniecky RI, Jackson GD, et al. (2005) A developmental and genetic classification for malformations of cortical development. Neurology 65: 1873-1887.
- Barkovich AJ, Guerrini R, Kuzniecky RI, et al. (2012) A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 135: 1348-1369.
- Widdess-Walsh P, Kellinghaus C, Jeha L, et al. (2005) Electro-clinical and imaging characteristics of focal cortical dysplasia: Correlation with pathological subtypes. Epilepsy Res 67: 25-33.
- Kakita A, Kameyama S, Hayashi S, et al. (2005) Pathologic features of dysplasia and accompanying alterations observed in surgical specimens from patients with intractable epilepsy. J Child Neurol 20: 341-350.
- Colombo N, Tassi L, Galli C, et al. (2003) Focal cortical dysplasias: MR imaging, histopathologic, and clinical correlations in surgically treated patients with epilepsy. AJNR Am J Neuroradiol 24: 724-733.
- Kim DW, Kim S, Park SH, et al. (2012) Comparison of MRI features and surgical outcome among the subtypes of focal cortical dysplasia. Seizure 21: 789-794.
- Worrell GA (2012) Imaging focal cortical dysplasia in refractory epilepsy. Epilepsy Curr 12: 35-36.
- Seifer G, Blenkmann A, Princich JP, et al. (2012) Noninvasive approach to focal cortical dysplasias: Clinical, EEG, and neuroimaging features. Epilepsy Res Treat.
- Boonyapisit K, Najm I, Klem G, et al. (2003) Epileptogenicity of focal malformations due to cortical development: Direct electrocortographic-histopathologic correlations. Epilepsia 44: 69-76.
- Morioka T, Nishio S, Ishibashi H, et al. (1999) Intrinsic epileptogenicity of focal cortical dysplasia as revealed by magnetoencephalography and electrocorticography. Epilepsy Res 33: 177-187.
- Akhtari M, Salamon N, Duncan R, et al. (2006) Electrical conductivities of the freshly excised cerebral cortex in epilepsy surgery patients; correlation with pathology, seizure duration, and diffusion tensor imaging. Brain Topogr 18: 281-290.
- Marusic P, Najm I, Ying Z, et al. (2002) Focal cortical dysplasias in eloquent cortex: Functional characteristics and correlation with MRI and histopathologic changes. Epilepsia 43: 27-32.
- Kirschstein T, Fernandez G, Grunwald T, et al. (2003) Heterotopias, cortical dysplasias, and glioneural tumors participate in cognitive processing in patients with temporal lobe epilepsy. Neurosci Lett 338: 237-241.
- Duchowny M (2009) Clinical, functional, and neurophysiologic assessment of dysplastic cortical networks: Implications for cortical functioning and surgical management. Epilepsia 50: 19-27.
- Blackmon K, Kuzniecky R, Barr W, et al. (2015) Cortical gray-white matter blurring and cognitive morbidity in focal cortical dysplasia. Cereb Cortex 25: 2854-2862.
- Cepeda C, Hurst RS, Flores-Hernandez J, et al. (2003) Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J Neurosci Res 72: 472-486.
- Cendes F, Cook MJ, Watson C, et al. (1995) Frequency and characteristics of dual pathology in patients with lesional epilepsy. Neurology 45: 2058-2064.
- Fauser S, Huppertz HJ, Bast T, et al. (2006) Clinical characteristics in focal cortical dysplasia: A retrospective evaluation in a series of 120 patients. Brain 129: 1907-1916.
- Ho SS, Kuzniecky RI, Gilliam F, et al. (1998) Temporal lobe developmental malformations and epilepsy: Dual pathology and bilateral hippocampal abnormalities. Neurology 50: 748-754.
- Eriksson SH, Nordborg C, Rydenhag B, et al. (2005) Parenchymal lesions in pharmacoresistant temporal lobe epilepsy: Dual and multiple pathology. Acta Neurol Scand 112: 151-156.
- Mueller SG, Laxer KD, Cashdollar N, et al. (2006) Spectroscopic evidence of hippocampal abnormalities in neocortical epilepsy. Eur J Neurol 13: 256-260.
- Cascino GD, Trenerry MR, So EL, et al. (1996) Routine EEG and temporal lobe epilepsy: Relation to long-term EEG monitoring, quantitative MRI, and operative outcome. Epilepsia 37: 651-656.
- Fauser S, Schulze-Bonhage A (2006) Epileptogenicity of cortical dysplasia in temporal lobe dual pathology: An electrophysiological study with invasive recordings. Brain 129: 82-95.
- Lee SK, Kim DW (2013) Focal cortical dysplasia and epilepsy surgery. J Epilepsy Res 3: 43-47.
- French JA, Williamson PD, Thadani VM, et al. (1993) Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol 34: 774-780.
- Davies K, Hermann B, Dohan FC, et al. (1996) Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 24: 119-126.
- Park K, Chu K, Jung K, et al. (2010) Role of cortical dysplasia in epileptogenesis following prolonged febrile seizure. Epilepsia 51: 1809-1819.
- Annegers JF, Hauser WA, Shirts SB, et al. (1987) Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 316: 493-498.
- Brekelmans GJF, vanEmde Boas W, Velis DN, et al. (1995) Mesial temporal versus neocortical temporal lobe seizures: Demonstration of different electroencephalographic spreading patterns by combined use of subdural and intracerebral electrodes. J Epilepsy 8: 309-320.
- Tassi L, Garbelli R, Colombo N, et al. (2010) Type I focal cortical dysplasia: Surgical outcome is related to histopathology. Epileptic Disord 12: 181-191.
- Bower SP, Kilpatrick CJ, Vogrin SJ, et al. (2000) Degree of hippocampal atrophy is not related to a history of febrile seizures in patients with proved hippocampal sclerosis. J Neurol Neurosurg Psychiatry 69: 733-744.
- Wiebe S, Blume W, Girvin J, et al. (2001) A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 345: 311-318.
- Hudgins RJ, Flamini JR, Palasis S, et al. (2005) Surgical treatment of epilepsy in children caused by focal cortical dysplasia. Pediatr Neurosurg 41: 70-76.
- Kloss S, Pieper T, Pannek H, et al. (2002) Epilepsy surgery in children with focal cortical dysplasia (FCD): Results of long-term seizure outcome. Neuropediatrics 33: 21-26.
- Kral T, Clusmann H, Blumcke I, et al. (2003) Outcome of epilepsy surgery in focal cortical dysplasia. J Neurol Neurosurg Psychiatry 74: 183-188.
- Wyllie E, Comair YG, Kotagal P, et al. (1998) Seizure outcome after epilepsy surgery in children and adolescents. Ann Neurol 44: 740-748.
- Xue H, Cai L, Dong S, et al. (2016) Clinical characteristics and post-surgical outcomes of focal cortical dysplasia subtypes. J Clin Neurosci 23: 68-72.
- Rowland NC, Englot DJ, Cage TA, et al. (2012) A meta-analysis of predictors of seizure freedom in the surgical management of focal cortical dysplasia. J Neurosurg 116: 1035-1041.
- Ramırez-Molina JL, Di Giacomo R, Mariani V, et al. (2017) Surgical outcomes in two different age groups with focal cortical dysplasia type II: Any real difference? Epilepsy Behav 70: 45-49.
- Fauser S, Schulze-Bonhage A, Honegger J, et al. (2004) Focal cortical dysplasias: surgical outcome in 67 patients in relation to histological subtypes and dual pathology. Brain 127: 2406-2418.
- Isler C, Kucukyuruk B, Ozkara C, et al. (2017) Comparison of clinical features and surgical outcome in focal cortical dysplasia type 1 and type 2. Epilepsy Res 136: 130-136.
- Salanova V, Markand O, Worth R (2004) Temporal lobe epilepsy: Analysis of patients with dual pathology. Acta Neurol Scand 109: 126-131.
- Luan L, Sun Y, Yang K (2018) Surgical strategy for temporal lobe epilepsy with dual pathology and incomplete evidence from EEG and neuroimaging. Exp Ther Med 16: 4886-4892.
- Li LM, Cendes F, Andermann F, et al. (1999) Surgical outcome in patients with epilepsy and dual pathology. Brain 122: 799-805.
- Wagner J, Urbach H, Niehusmann P, et al. (2011) Focal cortical dysplasia type IIb: Completeness of cortical, not subcortical, resection is necessary for seizure freedom. Epilepsia 52: 1418-1424.
- Luders H, Najm I, Nair D, et al. (2006) The epileptogenic zone: General principles. Epileptic Disord 8: S1-S9.
- Fauser S, Haussler U, Donkels C, et al. (2013) Disorganization of neocortical lamination in focal cortical dysplasia is brain-region dependent: Evidence from layer-specific marker expression. Acta Neuropathol Commun 1: 1-13.
- Hyslop A, Miller I, Bhatia S, et al. (2015) Minimally resective epilepsy surgery in MRI-negative children. Epileptic Disord 17: 263-274.
- Jayakar P, Gaillard WD, Tripathi M, et al. (2014) Diagnostic test utilization in evaluation for resective epilepsy surgery in children. Epilepsia 55: 507-518.
Corresponding Author
Brandon M Korman, Ph.D, Psy D, Nicklaus Children's Hospital Brain Institute, Herbert Wertheim School of Medicine, Florida International University, 3100 SW 62nd Ave., Miami, Florida 33155, USA, Tel: 305-666-6511, ext 2469, Fax: 305-663-6857.
Copyright
© 2019 Korman BM. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.