A 6-Month-Old Boy with Congenital Hydrocephalus and Syndactylia Deformity
Abstract
Background
Craniosynostosis syndromes are rare and prognosis would benefit from early diagnosis and treatment. We tried to establish a fast molecular diagnosis method to detect craniosynostosis syndromes.
Methods
One patient aged 6 months was observed and diagnosed at first, and then another 10 patients were included in this study. All of them suffered from congenital hydrocephalus with normal head circumference. Karyotype analysis, chromosome genome microarray analysis and whole exome sequencing were used.
Results
The first patient was diagnosed as Apert Syndrome based on his clinical features and whole exome sequencing. The other 10 patients were also detected by the same protocol and one of them was found to be with Crouzon Syndrome. Both of the patients with craniosynostosis syndrome were treated in time and benefited from this new diagnosis protocol.
Conclusions
Craniosynostosis syndromes cause hearing loss and deformity. It is worthy to detect mutations on FGFR1, FGFR2 and FGFR3 for children who suffer from congenital hydrocephalus with normal head circumference.
Keywords
Craniosynostosis syndrome, Hydrocephalus, Diagnosis
Abbreviations
SD: Standard Deviation; CSF: Cerebrospinal Fluid; CT: Computed Tomography; DQ: Developmental Quotient
Case Description
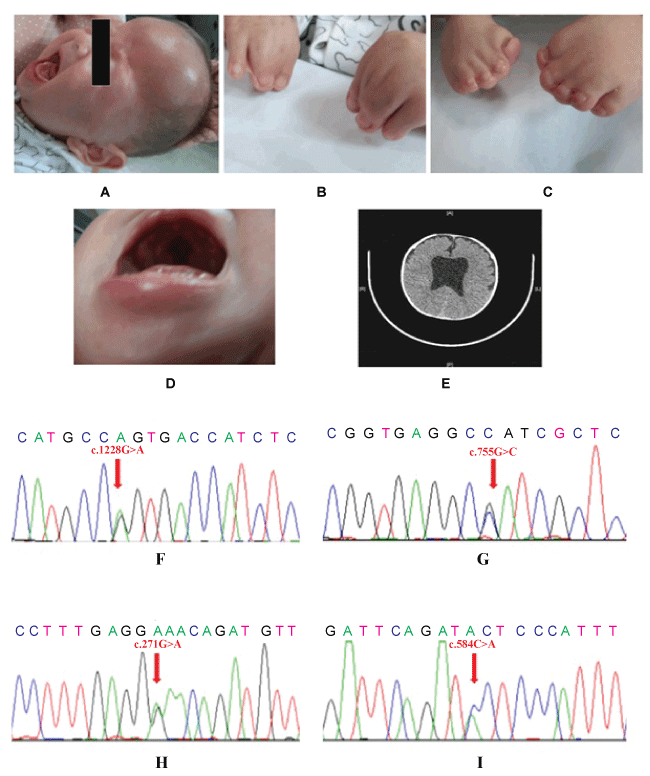
A 3.2 (correct writing form of weight is 3200 gr) kg boy was born with syndactylia deformity in November 2013. The Apar (Apgar) scores were 9.0 and 10 at post-delivery 1st, 5th minute, respectively, with no birth injury. His head circumference was 36.3 cm (+ 2SD) [1] and bregma was plump, suspicion of suffering with congenital hydrocephalus. After six months, the child was 6.0 kg in weight, 66 cm in height, 42 cm (-1SD) [1] of head circumference, 3 cm × 3 cm of anterior fontanelle and apophysis, no setting-sun sign, syndactylia deformity occurs both in bilateral fingers and toes, cleft palate (Figure 1A, Figure 1B, Figure 1C and Figure 1D). All the tests of Cerebrospinal Fluid (CSF) for viruses, bacteria and parasites identification showed negative results. Computed Tomography (CT) scan illustrated bilateral lateral ventricle enlargement (Figure 1E).
Parts of patients with congenital hydrocephalus could have an abnormal increase of head circumference caused by intracranial pressure mismatching body development, and the patient was with head circumference in the normal range [1] even intracranial hypertension. There were excessive hydrocephalus in ventricle and hydrocephalus obviously resulted in intracranial hypertension (Figure 1A and Figure 1E). It seemed that the patient's head circumference had not been influenced by hydrocephalus alone, and there simultaneously were with other body deformities. Syndrome should be taken into account.
We performed karyotype analysis and chromosome genome microarray analysis (HD Cytoscan, Affymetrix Co. Ltd, USA) for this patient at its initial diagnostics. However, there were no positive findings. After the discussion with the parents to explain the process and purpose of whole exome sequencing, we then applied the whole exome sequencing to detect the patient and his parents' DNA. There were 18 mutations found in the patient's exome and only 4 de novo DNA mutations were found, through comparing with his parents, who do not have any symptoms, and verified by Sanger DNA sequencing (Table 1, Figure 1F, Figure 1G, Figure 1H and Figure 1I).
Patient Follow-up
The syndrome should be attributed to the mutation on FGFR2, which FGFR2 mutations could result in several subtypes of craniosynostosis syndrome, such as Crouzon syndrome, Apert syndrome, Pfeiffer syndrome and so on [2].
FGFR-related craniosynostosis syndrome is complex genetic disorders with clinically distinct phenotypes, although most mutations exist on FGFR1, FGFR2 and FGFR3 genes. The clinical features of Crouzon syndrome, Apert syndrome and Pfeiffer syndrome are referred [FGFR-Related Craniosynostosis Syndromes. Gene Reviews®. http://www.ncbi.nlm.nih.gov/books/NBK1455/ (Last update June 2011)]. Based on the symptoms of this patient: normal weight and height, hydrocephalus, syndactylia deformity of all the fingers and FGFR2c.755 G > C mutation, all of which are closely related to Apert syndrome. Therefore, the diagnosis of this patient should be Apert syndrome, whose incidence is about 1 in 65,000 live births [3].
There is an odds of 50% to result in intellectual disability for Apert syndrome and drainage could reduce intracranial hypertension to improve the outcome by hydrocephalus surgery if there was no irreversible mental damage had appeared. Gesell development scale test was applied when the patient was six-month-old and the DQ value was 24.4, showing that the intelligence development level of the patient was significantly lower than that of normal children. Since it had caused serious and irreversible mental damage, it was not necessary of hydrocephalus surgery. The incidence of hearing loss in Apert syndrome is nearly 80%, mainly because of skull malformation [4]. Therefore, a comprehensive hearing test was performed and hearing was normal. The patient was advised to receive surgery for cleft palate at the age of one-year.
The patient is alive until August, 2015. Fundoscopic exam and hearing evaluation (pure tone audiometry, acoustic immitance test and auditory brainstem response) were routinely performed and no positive findings. The hydrocephalus symptom is not improved or deteriorated. Therefore, the patient has not received surgery for cleft palate.
Case Discussion
This case represented a normal head circumference even an intracranial hypertension caused by hydrocephalus. When a patient is with hydrocephalus and deformity of other organs, it reminds us that the patient is with a syndrome (like Apert or Pfeiffer). If patients do not have any obvious deformities (like Crouzon), it is risky to diagnose them as hydrocephalus alone.
Besides, we also collected 10 patients (7 boys and 3 girls) who were suffered from congenital hydrocephalus with normal head circumferences. All the tests of CSF for viruses, bacteria and parasites identification showed negative results. There was no deformity found among them. Peripheral blood from the 10 patients was collected and DNA was extracted. Each exon of FGFR1, FGFR2 and FGFR3 genes were amplified by PCR and analyzed by DNA Sanger sequence. One of them with low-set ears was found to be with p.S252W mutation on FGFR2 and another patient with strabismus was found to be with p.S347C mutation on FGFR2 [2,5]. There were no syndactylia deformity of their fingers or toes, and no evidence to support mental retardation, whose symptoms were related to Crouzon syndrome. If they were not analyzed by DNA sequence, they would have been diagnosed as simple congenital hydrocephalus. Both of them were received hearing test in view of high incidence of hearing loss caused by Crouzon syndrome [4]. It is helpful to screen FGFR genes to check the children who suffer from congenital hydrocephalus with normal head circumference. It is helpful to diagnose craniosynostosis syndrome, and their prognosis would benefit from reasonable diagnosis.
Acknowledgements
We thank all patients and guardians involved in the study.
Authors' Disclosures or Potential Conflicts of Interest
No authors declared any potential conflicts of interest.
Patient Consent
Obtained.
References
- Zong XN, Li H (2013) Construction of a new growth references for China based on urban Chinese children: comparison with the WHO growth standards. PLoS One 8: e59569.
- Passos-Bueno MR, Richieri-Costa A, Sertié AL, et al. (1998) Presence of the Apert canonical S252W FGFR2 mutation in a patient without severe syndactyly. J Med Genet 35: 677-679.
- Fearon JA (2003) Treatment of the hands and feet in Apert syndrome: an evolution in management. Plast Reconstr Surg 112: 1-12.
- Agochukwu NB, Solomon BD, Muenke M (2014) Hearing loss in syndromic craniosynostoses: otologic manifestations and clinical findings. Int J Pediatr Otorhinolaryngol 78: 2037-2047.
- Shotelersuk V, Mahatumarat C, Ittiwut C, et al. (2003) FGFR2 mutations among Thai children with Crouzon and Apert syndromes. J Craniofac Surg 14: 101-104.
Corresponding Author
Prof. Ping Liang, Department of Neurosurgery, Ministry of Education Key Laboratory of Development and Disorders, Key Laboratory of Pediatrics in Chongqing, Children's Hospital, Chongqing Medical University, China;
Prof. Lin Zou, Center for Clinical Molecular Medicine, Ministry of Education Key Laboratory of Development and Disorders, Key Laboratory of Pediatrics in Chongqing, Children's Hospital, Chongqing Medical University, China.
Copyright
© 2017 Liu Z, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.