Hepatopulmonary Syndrome Due to Congenital Extrahepatic Portosystemic Shunts: A Case Report
Abstract
Congenital extra hepatic port systemic shunt, is a rare clinical entity which can lead to hepatopulmonary syndrome wherein blood from portal vein directly drains into systemic circulation, causing an alteration in metabolism of pulmonary vasoactive substances, pulmonary vasodilatation, diffusion-perfusion defects, and eventually, arterial hypoxemia due to either an increase in pulmonary vascular resistance or the creation of arteriovenous malformations. We report a six-year-old male presented with a 3-year history of cyanosis and dyspnea. Initially the diagnosis of cystic fibrosis was initially suspected, but later ruled out. He was ultimately diagnosed with primary pulmonary hypertension through cardiac catheterization for which he was referred to our center after deteriorating clinically.
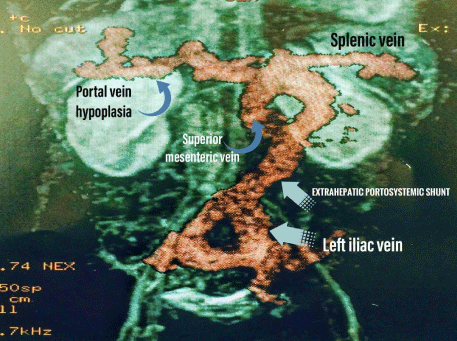
The patient exhibited a decrease in arterial pO2 by 8% with change in position from decubitus-supine to standing. Abdominal ultrasound, triple-phase CT scan and MRI all confirmed the portocaval, extra hepatic, type II shunt though visualization of the thickened collaterals emptying into the left iliac vein. Surgical ligation of the shunt near the portal vein was undertaken. As an outpatient, the patient saturated 95% without the need for supplemental oxygen. Portal anatomy and intrahepatic flow improved and are now considered normal. We : Must consider the diagnosis of congenital extra hepatic port systemic shunt, in children presenting with cyanosis and unspecific liver, cardiac or pulmonary disease. These patients are at high risk for developing hepatic encephalopathy, hepatopulmonary syndrome and portal hypertension. As such, closure of the shunt should be considered early in Abernethy malformation.
Keywords
Porto systemic, Shunt, Abernethy, Hepatopulmonary
Introduction
Hepatopulmonary syndrome (HPS) is a triad of liver disease, arterial hypoxemia, and pulmonary vascular dilatation [1]. Congenital portosystemic shunts (CPSS) are one of the causes of hepatopulmonary syndrome [1,2]. Arterial hypoxemia is caused by functional intrapulmonary right-to-left shunt secondary to pulmonary vascular dilatation at the capillary and pre-capillary levels [3].
CPSS are classified into intrahepatic and extra hepatic shunts. Extrahepatic shunts, also known as Abernethy malformation (AM), are characterized by extra hepatic congenital diversion of portal blood away from the liver [1]. As of 2010, 83 cases of AM have been reported since it was first described in 1793 by John Abernethy [2- 4]. Morgan and Superina, proposed in 1994 the classification of extra hepatic port systemic anomalies, intrahepatic shunts were first classified by Park in 1990. In complete extra hepatic shunts the anastomoses are established end to side between the tributaries of the portal or mesenteric system (Type 1a) or main portal vein (PV) as a trunk (Type 1b) and a systemic vein. In both intrahepatic PV branches are absent. In partial shunt, (Type 2) PV radicles are present, however, there is diversion of the portal blood into a systemic vein through a side-to-side or end-to-side shunt [5-8].
As a result of CPSS, the blood from spleen and the intestines drain into inferior vena cava (IVC) through a shunt bypassing the liver, thereby causing an alteration in metabolism of pulmonary vasoactive substances, pulmonary vasodilatation, diffusion-perfusion defects, and eventually, arterial hypoxemia due to either an increase in pulmonary vascular resistance or the creation of arteriovenous malformations. The ensuing shunting of unmetabolized toxic substances in the postprandial state, from the mesenteric circulation into the systemic circulation via IVC leads to varying degrees of encephalopathy [1-9]. There is a wide spectrum of therapeutic options for CPSS, ranging from noninvasive conservative treatment to liver transplantation [10]. We describe a case of extra hepatic type 2 shunt, managed surgically with a satisfactory postoperative course.
It is very important to remark that pediatric patients presenting with structurally normal echocardiograms and unexplained cyanosis should be worked up to rule out the possibility of a CPSS with HPS, even if the patient has no previous history of cardiac or hepatic illness.
Case Description
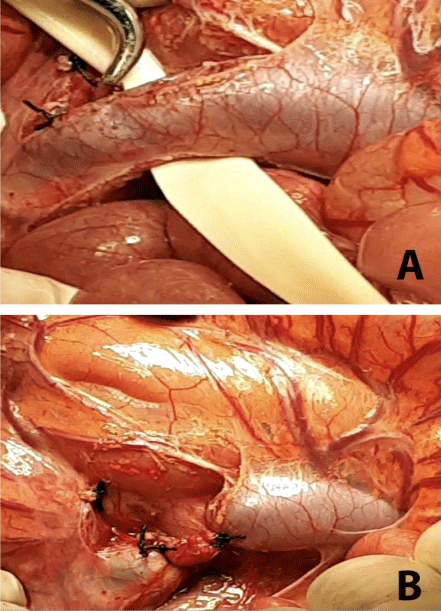
A six-year-old male presented with a 3-year history of cyanosis and dyspnea. Initially the diagnosis of cystic fibrosis was initially suspected, but later ruled out with diagnostic testing. He was ultimately diagnosed with primary pulmonary hypertension through cardiac catheterization for which he was referred to our center after deteriorating clinically. On admission, he presented with an SpO2 of 78% with an FiO2 of 80%. The patient had central and peripheral cyanosis, clubbing of the fingers, somnolence, breath smelling of fish, and low weight for his height. His bilirubin, transaminases and alkaline phosphates were all slightly elevated. Chest x-ray showed signs of pulmonary congestion. Echocardiogram demonstrated no septal defects, but signs of mild pulmonary hypertension. An Echocardiogram bubble study confirmed the passage of bubbles from the right to the left chambers after the fourth beat. The patient exhibited a decrease in arterial pO2 by 8% with change in position from decubitus-supine to standing. Abdominal ultrasound, triple-phase CT scan and MRI all confirmed the portocaval, extrahepatic, type II shunt though visualization of the thickened collaterals emptying into the left iliac vein (Figure 1) Surgical ligation of the shunt near the portal vein was undertaken (Figure 2).
On postoperative day 10, the patient became thrombocytopenic and experienced one episode of desaturation. The patient was diagnosed with bilateral pulmonary infiltrates by chest x-ray. Altogether this led to the diagnosis of platelet sequestration within pulmonary arteriovenous fistulas. The patient was managed with anticoagulation with low-molecular weight heparin. Postoperatively, pulmonary artery pressure increased to 80 mmHg, which responded to sildenafil 2 mgxkg over the course of one year. As an outpatient, the patient saturated 95% without the need for supplemental oxygen. Portal anatomy and intrahepatic flow improved, after two year of follow up, are considered normal (Figure 3). The patient’s functional capacity and prognosis are excellent.
Discussion
In the setting of hepatic diseases, liver dysfunction or high portal pressure have been thought to contribute to the pathogenesis of HPS. (1) Three hypothesis accounting for the etiology have been proposed, including: (i) elevated ET-1 (endothelin-1) circulating throughout the body leading to an up regulation in nitric oxide (NO) production in the lungs via continuous stimulation of NO synthase, (ii) hepatic products necessary for pulmonary vasomotor control are decreased by liver dysfunction or hepatic venous flow reduction, (iii) translocation of gut bacteria, leading to an activation of alveolar macrophages resulting in increase in inducible NO synthase [11].
HPS was first described in a patient with Abernethy malformation by Alvarez et al [12]. More than 65 % patients with extra hepatic CPSS are females and more than 22 % of patients have a concomitant congenital heart disease [7]. These are postulated to develop due to the close relationship between the development of the heart and the vitelline veins in embryonic development. Congenital anomalies of the portal venous system result from abnormal coalescence of the vitello-umbilical venous plexus during embryogenesis. These rare anomalies include cavernous transformation of the portal vein, preduodenal portal vein or peribiliary portal vein, duplication of portal vein, and portal vein atresia [2].
Other associated anomalies commonly found include those of the spleen, urinary and male genital tract, brain and skeleton. Hepatic changes such as focal nodular hyperplasia, hepatocellular carcinoma, and hepatoblastoma are diagnosed in more than 40 % of patients. These anomalies are less common in patients in type 2 shunts [2, 6-7]. The original report by Abernethy included findings of right-sided heart and right aortic arch [1].
CPSS symptomatology includes jaundice, difficulty in breathing, cyanosis, clubbing, and abdominal mass. Patients frequently are generally worked up for cardiac and pulmonary shunts. Hypoxemia leads to neoangiogenesis and vascular hyperplasia which in turns leads to digital clubbing. It has also been postulated that damaged liver metabolites fail to break down substances responsible for the local changes in bone metabolism in these patients [1,6]. CPSS are often detected early in countries that screen galactose levels in newborns [13]. Other symptoms related to the shunts/hepatic dysfunction include pulmonary hypertension, hyperandrogenism, primary amenorrhea, signs of virilization and encephalopathy [14-16]. Prior percutaneous liver biopsy leading to intrahepatic port systemic venous shunt is a common intrahepatic cause, and should always be excluded before diagnosing a case as congenital [17].
The diagnosis of CPSS is most frequently established with ultrasound. Additional studies including multidetector computed tomography angiography, magnetic resonance imaging, portovenography and scintigraphy, will show the shunt, the presence of any intrahepatic PV branches and pulmonary vasculature; these studies are also useful in the assessment of accompanying liver tumors and malformations [1-3,10,12].
The treatment of CPSS includes dietary control (restriction of protein, ingestion of lactulose, oral administration of no absorbable antibiotics) and surgery. Symptomatic patients with extra hepatic shunt, that is those with port systemic encephalopathy, liver dysfunction, or shunt ratio > 60 %, are divided into two groups according to the type of shunt. Those with type 1 shunts should be transplanted, whereas those with type 2 shunts should be offered shunt closure-either through mobilization with interventional radiology or surgical intervention. Patients with type 1 shunts may not be offered shunt occlusion because it represents the only route for intestinal and splenic blood drainage [10,18-20].
It is also recommended that type 2 shunts should definitely be closed in asymptomatic patients in order to prevent the development of complications at a later date. It has been reported that hypo plastic PV branches are able to expand after the closure of shunt. This occurred in our patient in this case. Shunt closure results in restoration of intrahepatic portal blood flow [2,16,20]. Another option in both types of CPSS are angiographic intervention with coil embolization [17,20].
Although diagnosis of these malformations is unusual, one must consider the diagnosis of CPSS in children presenting with cyanosis and unspecific liver, cardiac or pulmonary disease. Doppler echocardiography, CT angiography and MRI are the best diagnostic methods. These patients are at high risk for developing hepatic encephalopathy, hepatopulmonary syndrome and portal hypertension. As such, closure of the shunt should be considered early in Abernethy malformation. Clinical regression of symptomatology and stabilization of pulmonary, cardiac, neurological, and renal complications is seen following shunt closure.
Declaration
Consent for publication: Written informed consent was obtained (from the parents as patient is 6 year old) for publication of this case report and accompanying images in spite that the patient can not be identified.
Conflict of Interests
The authors declare that they have no conflict of interests.
Financial Support
No.
Author Constribution
All authors read and approved the final manuscript.
Ethical Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). Written informed consent was obtained from the patient.
References
- Sahu MK, Bisoi AK, Chander NC, et al. (2015) Abernethy syndrome, a rare cause of hypoxemia: A case report. Ann Pediatr Card 8: 64-66.
- Mistinova J, Valacsai F, Varga I et al. (2010) Congenital absence of the portal vein-case report and a review of literature. Clin Anat 23: 750-758.
- Krowka MJ (2000) Hepatopulmonary syndrome. Gut 46: 1-4.
- Abernethy J (1793) Account of two instances of uncommon formation, in the viscera of the human body. Philos Trans R Soc Lond B Biol Sci 83: 59-66.
- Morgan G, Superina R (1994) Congenital absence of the portal vein: Two cases and a proposed classification system for portasystemic vascular anomalies. J. Pediatr. Surg. 29: 1239-1241.
- Ghuman S S, Gupta S, Buxi T, et al. (2016) The abernethy malformation-myriad imaging manifestations of a single entity. Indian J Radiol Imaging 26: 364-372.
- Avila LF, Luis AL, Encinas JL, et al. (2006) Shunt porto cava congénito. Malformation de Abernethy. Cir Pediatr 19: 204-209.
- Park JH, Cha SH, Han JK, et al. (1990) Intrahepatic portosystemic venous shunt. AJR Am J Roentgenol 155: 527-528.
- Doraiswamy V, Sivakumar K (2017) Impact of Abernathy malformation on pulmonary circulatory hemodynamics in a univentricular heart. Ann Pediatr Card 10: 90-91.
- Ringe K, Schirg E, Melter M, et al. (2008) Congenital absence of the portal vein (capv). two cases of abernethy malformation type 1 and review of the literature. Radiologe 48: 493-502.
- Rabiller A, Nunes H, Lebrec D, et al. (2002) Prevention of gram negative translocation reduces severity of hepatopulmonary syndrome. Am J Respir Crit Care Med 166: 514-517.
- Alvarez AE, Ribeiro AF, Hessel G, et al. (2002) Abernethy malformation: One of the etiologies of hepatopulmonary syndrome. Pediatr Pulmonol 34: 391-394.
- Ono H, Mawatari H, Mizoguchi N, et al. (1998) Clinical features and outcome of eight infants with intrahepatic porto-venous shunts detected in neonatal screening for galactosaemia. Acta Paediatr 87: 631-634.
- Satoh M, Yokoya S, Hachiya Y, et al. (2001) Two hyperandrogenic adolescent girls with congenital portosystemic shunt. Eur J Pediatr 160: 307-311.
- Wakamoto H, Manabe K, Kobayashi H, et al. (1999) Subclinical portal-systemic encephalopathy in a child with congenital absence of the portal vein. Brain Dev 21: 425-428.
- Franchi-Abella S, Branchereau S, Lambert V, et al. (2010) Complications of congenital port systemic shunts in children: Therapeutic options and outcomes. J Pediatric Gastroenterology Nutr 51: 322-330.
- Saxena AK, Sodhi KS, Arora J , et al. (2004) Congenital Intrahepatic Porto systemic Venous Shunt in an Infant with Down Syndrome. AJR 183:1783-1784.
- Gallego C, Miralles M, Marín C, et al. (2004) Congenital hepatic shunts. Radiographics 24: 755-772.
- Akahoshi T, Nishizaki T, Wakasugi K, et al. (2000) Portal-systemic encephalopathy due to a congenital extrahepatic portosystemic shunt: Three cases and literature review. Hepatogastroenterology. 47: 1113-1116.
- Vicente N, Pérez M, Gander R, et al. (2015) Shunt portosistémico congénito. Importancia del tratamiento precoz. Cir Pediatr 28: 67-73.
Corresponding Author
Dr. Luis Marcano, Chief of Pediatric Cardiothoracic Surgery Unit, Vicente Corral Moscoso, Ecuador; Tel: 593-998588878.
Copyright
© 2022 Miurkis E, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.