Unusual Diagnosis and Extended Management of Chronic Inflammatory Demyelinating Polyneuropathy in an Unimmunized Patient
Abstract
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) is a rare autoimmune disorder, often of idiopathic cause, but one that can result as a complication of Guillain Barre Syndrome (GBS). It often affects patients around 50-years of age, and impacts males twice as much as females. The treatment plan and management of the disease is still being investigated and developed. Our report describes findings consistent with CIDP in a pediatric patient, with an undiagnosed infectious prodrome. The goals of this case report are to increase knowledge to correctly diagnose these patients that may have an atypical presentation in the pediatric population and to add to the data for longitudinal management of the disease.
Background
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) is a rare autoimmune disorder, often of idiopathic cause but that can result as a complication of Guillain Barre Syndrome (GBS). It is characterized by hyporeflexia or areflexia with typically, descending weakness causing pure motor deficits (proximal and distal), although pure sensory or mixed deficits can also be seen. It often presents in middle-aged adults in their 50's with males affected twice as much as females. The incidence among adults is 1-2 cases per 100,000 people. However, CIDP incidence rate in patients under age 20 is 0.48 per 100,000 and the distal variant is more commonly encountered [1,2]. The diagnostic work-up and preferred treatment strategy for pediatric patients is similar to that of the adult population, as outlined by the European Federation of Neurological Societies/Peripheral Nerve Society Guideline in 2010 [3]. However, presentation of the disease in the pediatric population varies slightly from that of the adult population and pure motor symptoms are more often encountered. In this case, we aim to highlight this atypical presentation of CIDP and the further need for more recent guidelines to help with long term treatment for CIDP in pediatric patients.
Case
An 8-year-old unvaccinated female presented to Joe DiMaggio Emergency Department on November 2021 with chief complaints of stiffening of lower extremities, increased tendency to fall, and muscle weakness. The symptoms started one month prior to presentation and there was no intercurrent illness or injury preceding onset of symptoms. Patient was otherwise healthy, had no known sick-contacts or COVID-19 exposure and her vaccine immunizations were not up to date. She continued with normal PO intake and urine output. She was afebrile on presentation and her physical exam was significant for abnormal gait with a pelvic tilt to the left side, absent patellar reflexes, and 1+ Achilles reflexes bilaterally, weakness of proximal lower extremity muscle groups, and scattered abrasions over the anterior shins and knees. Original laboratory workup in the Emergency Department (ED) included CK, troponin, CMP, CBC, urinalysis which were unremarkable. CT brain and lumbar X-rays were also obtained and found to be unremarkable.
The patient was admitted to the hospital for further workup and subsequent MRI imaging of the spine with and without contrast was significant for thickening and enhancement of the cauda equina, possibly suggestive of Guillain Barre Syndrome (GBS). Lumbar puncture was performed revealing cytoalbuminologic dissociation, as seen in Table 1, further suggestive of GBS. Given the history, clinical exam, CSF and MRI findings, the patient was diagnosed with GBS and treated with IVIG 2 G/kg administered as daily infusions over three days. The patient complained of hip pain and mild headaches during IVIG treatments, but symptoms subsided and clinically her gait was significantly improved after the third dose of IVIG. The patient was discharged six days after presentation with instructions to follow up with her primary pediatrician, the neurology team, and outpatient physical therapy.
Comparison of cerebrospinal fluid (CSF) cell count with differential results from the patient's initial and second hospital admission. Both CSF results show isolated elevation of total protein consistent with albumin cytological dissociation, which is reflective of widespread inflammation of nerve roots. This CSF finding is classically associated with Guillain-Barre Syndrome.
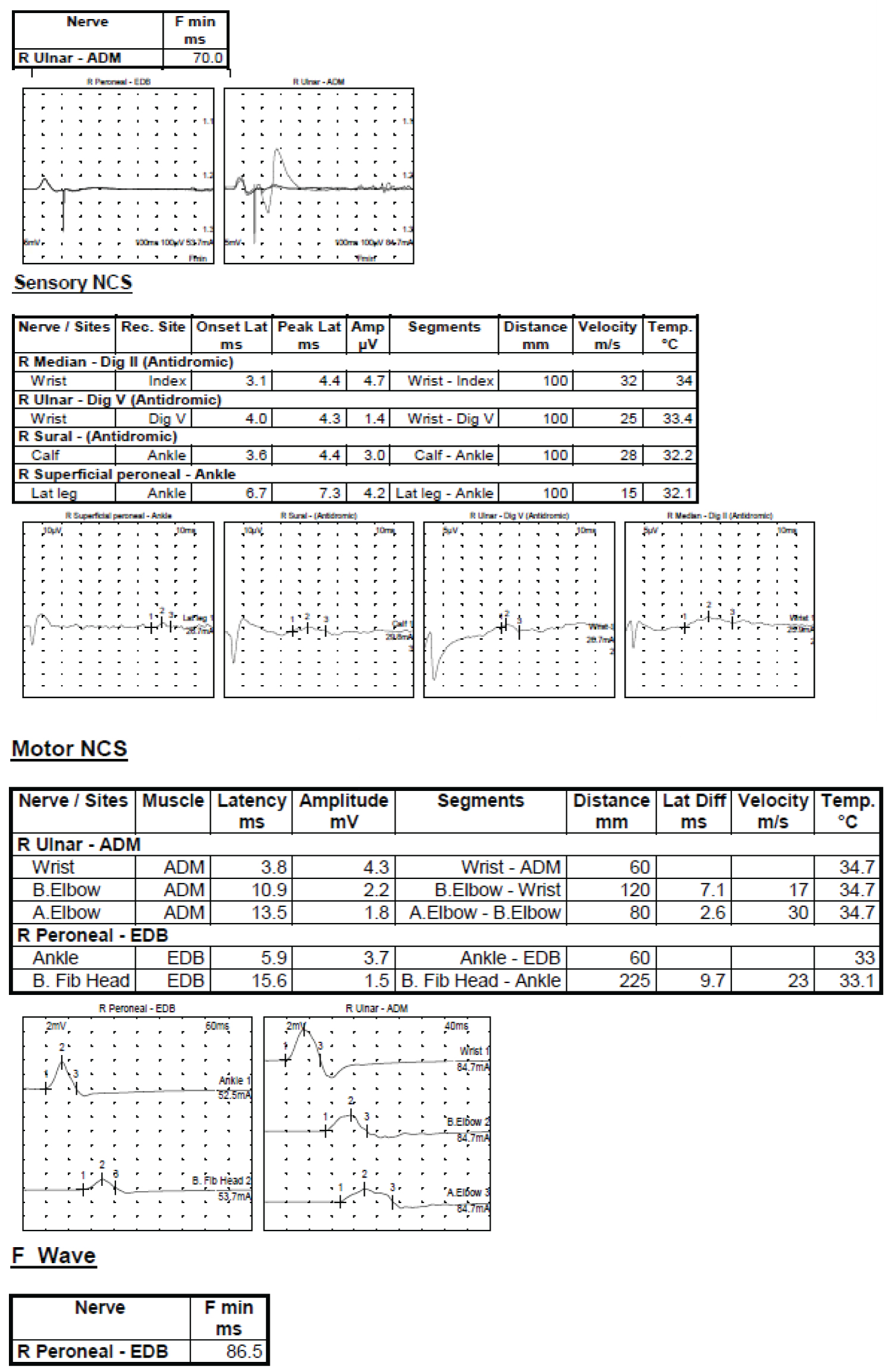
Less than three months after her initial hospitalization, the patient presented again in February 2022, for recurrence of symptoms over four days. She complained of gait instability, muscle weakness, and stuttering. The patient never followed up with the recommended outpatient providers due to the winter holiday season and it is unclear if her symptoms from initial presentation had completely resolved the day of presentation in February, the patient's muscle weakness and unsteadiness was significant enough that she was sent to the school nurse and her mother was contacted to pick her up from school. Her mother contacted the neurologist and was recommended to return to the ED. On presentation to the ED, the patient's physical exam was significant for weak hand grip, 4/5 strength in hips and ankles bilaterally, absent deep tendon reflexes bilaterally, altered gait with minimal hip flexion, modified Gower sign, and an inability to tiptoe, walk on her heels, or run with her arms extended. The working differential for her condition was expanded to include CIDP and the necessary studies were done to evaluate the diagnosis as per the European Joint task force diagnosis guidelines [3]. A repeat of the brain and spine imaging was not significant and a repeat lumbar puncture, as seen in Table 1, again found elevated protein. A cytokine panel was obtained which showed elevated NK cells, indicative of a possible viral etiology for her symptoms. Electromyogram and nerve conduction studies were conducted, as shown in Figure 1, consistent with motor and sensory demyelinating polyneuropathy. The clinical picture of the patient, as well as the lumbar puncture and EMG studies served to diagnose the child with CIDP. Throughout her hospital admission, the patient's vitals were within normal limits, and she remained afebrile with adequate oral intake and urine output.
She was started on an IVIG regiment, as described in Łukawska, et al. 2021. The study indicated significant symptom remission with an initial IVIG dose of 2 g/kg over 5 days followed by a maintenance dose of 0.4-1.2 g/kg every 2-6 weeks along with a steroid treatment [4]. The patient was discharged from the hospital five days later, after the initial IVIG dose was completed, in February 2022. She had remarkable improvement of her leg strength and gait but was not symptom free. The patient's mother shared her personal concerns with frequent steroid use in the patient due to its side effects, so the decision was made to schedule her for monthly IVIG infusions only. She was scheduled for follow-up with the outpatient physical therapy rehabilitation clinic, the pediatric allergy and immunology clinic, and the pediatric neurology clinic as well as with her primary pediatrician.
Electrodiagnostic study showing generalized abnormalities consistent with motor and sensory demyelinating polyneuropathy. This pattern of electrodiagnostic along with the patient's prolonged clinical history is consistent with CIDP.
Findings:
1. The right ulnar motor nerve shows normal digital latency and CMAP amplitude, however, there is evidence of conduction block below and across the elbow with significant decrease in the CMAP amplitudes and conduction velocities at these two sited. The F-wave for the ulnar nerve was also more than double of the expected latency.
2. The right peroneal motor nerve shows prolonged distal latency with normal CMAP amplitude with evidence of conduction block while stimulating below the fibular head with decreased CMAP amplitude and decreased conduction velocity. The F-wave for this nerve also showed a significant delay in the latency of more than double the expected value.
3. The right median sensory nerve shows prolonged latency and decreased conduction velocity with normal SNAP amplitude.
4. The right ulnar sensory nerves show prolonged latency with decreased conduction velocity and decreased SNAP amplitude.
5. The right sural sensory nerves show prolonged latency with decreased conduction velocity and normal SNAP amplitude.
6. The right superficial peroneal nerves show significant delay of the latency with decreased conduction velocity and normal SNAP amplitude.
On follow-up, the patient's monthly IVIG has been modified to an increased frequency of every three weeks due to a decreasing interval between her relapses. In accordance with her mother's preferences, she has never received steroid doses. She also is being followed by occupational and physical therapy to help her recover strength and resume daily activity level. Monthly laboratory findings consistently show normal immunoglobulin levels, but an elevation in her basophils and eosinophils, as well as an elevation in her total protein (8 g/dL), AST levels (55-60 g/dL) and a decreased alkaline phosphatase (< 160 g/dL). To prevent injuries and allow for more consistent adherence to her treatment plan, she has been placed in a homeschooling program temporarily for one semester.
Discussion
The incidence of Chronic inflammatory demyelinating polyneuropathy is 1-2 per 1,00,000 in adults, and 0.23-0.48 per 1,00,000 in children [1,2]. The rarity of this disease coupled with the lack of definitive evidence on treatment strategies makes the disease complex to diagnose and treat. This case highlights the atypical presentation of CIDP in the pediatric population and the challenges of its treatment, requiring monthly, if not weekly, management and multiple modalities of treatment. We present an unvaccinated pediatric patient, who develops CIDP most likely following a viral infection months earlier. The patient had originally presented and been diagnosed with Guillain-Barre Syndrome, an acute form of demyelinating polyneuropathy, month after the viral infection, and while 95% of patients usually recover with varying degree of remission, some, like our patient, go on to develop the chronic form of the disease [5].
The diagnosis of CIDP as outlined by the European Federation of Neurological Societies/Peripheral Nerve Society Guideline in 2010 [3], involves a clinical picture consistent with the presentation of progressive and relapsing muscle weakness, either symmetric or asymmetric, developing over eight weeks and with absent or decreased deep tendon reflexes. Their guidelines also emphasize confirming the diagnosis with electrodiagnostic criteria (EMG), showing at least two motor nerve abnormalities. Other supportive criteria include a CSF analysis showing cytoalbuminologic dissociation, nerve biopsy or response to treatment.
The widely preferred treatment for CIDP has been IVIG 1-2 mg/kg daily or on alternate days [1]. It has been suggested to add steroids into CIDP regiment, however, studies have found that it was not as beneficial in pure motor CIDP [3,6]. Long-term treatment with steroids is also usually used with caution because of the adverse effect on a pediatric patient's growth and bone development. Another treatment option, as recommended by the European Joint Task force guidelines [6], includes the use of plasmapheresis in the case that IVIG and steroid treatment prove ineffective. Furthermore, they recommend adding immunosuppressants or immunomodulators if the maintenance dose is inadequate. The prognosis of CIDP in pediatric populations is generally good, with a remission rate of 51.4% and follow up period ranging from 10-222 months [4]. In our patient's case, steroids were avoided as maintenance therapy, and plasmapheresis was not started as an option since patient was responding to IVIG every 3 weeks. While the end-goal of her treatment is resolution of her symptoms, it is unclear at this time how aggressive the future treatment course will need to be and how long this presentation of CIDP will take to resolve.
In conclusion, more longitudinal studies will be useful in helping with consistent standard treatment strategies for pediatric CIDP.
Declaration
Ethics approval and consent to participate
This article does not contain any studies with human or animal subjects performed by any of the authors.
Consent for publication
Consent has been obtained from the legal guardian/parent.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due HIPAA, but are available from the corresponding author on reasonable request.
Competing interests
The authors of this manuscript declare no conflict of interest.
Funding
No funding was awarded or obtained for this project.
Authors contributions
Danan V: Active participant in the care of the patient, primary author of the manuscript; Fridman S: Author and editor of the manuscript; Lopez C: Active participant in the care of the patient, author and editor of the manuscript; Basit A: Primary physician of the patient, author and editor of the manuscript.
Acknowledgement
The authors would like to acknowledge and thank the patient and her family for allowing us to care for them and publish this interesting case report, Boston Children's Hospital for their consult, and Joe DiMaggio Children's Hospital for the support in publishing this manuscript.
References
- Markowitz JA, Jeste SS, Kang PB (2008) Child neurology: Chronic inflammatory demyelinating polyradiculoneuropathy in children. Neurology 71: e74-e78.
- Iijima M, Koike H, Hattori N, et al. (2008) Prevalence and incidence rates of chronic inflammatory demyelinating polyneuropathy in the Japanese population. J Neurol Neurosurg Psychiatry 79: 1040-1043.
- Van den Bergh PY, Hadden RD, Bouche P, et al. (2010) European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society - first revision. Eur J Neurol 17: 356-363.
- Łukawska M, Potulska-Chromik A, Lipowska M, et al. (2021) Pediatric CIDP: Diagnosis and Management. A Single-Center Experience. Front Neurol 12: 667378.
- https://www.uofmhealth.org/conditions-treatments/brain-neurological-conditions/guillain-barre-syndrome-GBS-and-chronic-inflammatory-demyelinating-polyneuropathy-CIDP
- Patel K, Bhanushali M, Muley SA (2010) Management strategies in chronic inflammatory demyelinating polyradiculoneuropathy. Neurol India 58: 351-360.
Corresponding Author
Victoria Danan, BA, Charles E. Schmidt College of Medicine, 777 Glades Road, BC-71, Boca Raton, FL 3343, USA
Copyright
© 2022 Danan V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.