A Novel PPP2CA Gene Variant Presenting with Dystonia and Progressive Brain Atrophy
Abstract
Introduction: PPP2CA gene mutations are rare and associated with a neurodevelopmental disorder with or without structural brain abnormalities. We present a case of a novel de novo PPP2CA gene mutation, namely c.176A > G (p.H is 59 Arg), leading to a phenotype of global developmental delay, seizures, dystonia and progressive brain atrophy.
Case report: The patient is a 3-year-old girl who presented to our Paediatric Neurology clinic at 10 weeks of age due to poor weight gain, gastro-oesophageal reflux disease, concerns about unsafe swallow and developmental delay. She was noted to have dysmorphic features and had developed seizures at the age of 2 months, later evolving into infantile spasms with hypsarrythmia on electroencephalography. Of note, she also suffered from persistent episodes with dystonic movements. MRI of the brain showed progressive global brain atrophy, a marked pontocerebellar overlay, focal signal changes in brain stem nuclei and globally delayed myelination. Whole genome trio DNA sequencing as part of the 100K genome project revealed a de novo heterozygous likely pathogenic PPP2CA gene variant: c.176A > G; (p.H is 59 Arg).
Since the diagnosis, the patient has unfortunately shown progressive reduction in head circumference, developmental regression, increasing seizure frequency and worsening of her dystonic symptoms.
Conclusion: We suggest that severe dystonia and progressive brain atrophy are features of this novel PPP2CA genetic variant, which causes the phenotype spectrum of this genetic mutation to expand.
Keywords
PPP2CA gene mutation, Dystonia, Progressive brain atrophy, Epileptic encephalopathy, Microcephaly
Introduction
Highly expressed in the brain, protein phosphatase 2A (PP2A) enzymes catalyse dephosphorylation of phospho-Ser and phosphor-The residues in a large variety of substrates, thereby counterbalancing Ser/The-specific protein kinases and playing a vital role in regulating cellular signalling [1]. PP2A holoenzymes comprise a catalytic C, scaffolding A- and regulatory B-type subunits [2]. These determine the substrate specificity, (sub)cellular localization and catalytic activity of the PP2A holoenzymes [1].
PP2A complexes are encoded by 19 different genes and de novo mutation in genes encoding specific subunits of type 2A protein phosphatases. Their links with intellectual disability (ID) and developmental delay (DD) have previously been documented [2-7]. It is only recently that mutations in both genes encoding the C subunit have been reported [2].
DNA variants in the PPP2CA gene are rare, with only one literature reference reporting 16 patients with PPP2CA gene variants [2]. However, there are reports of gene variants in PPP2RD5 and PPPP2R1A, genes which together with PPP2CA are all involved in PPP2A (Protein Phosphatase 2A) protein dysfunction.
The features associated with pathogenic PPP2CA variants reported in the literature are: Mild to profound ID and DD, severe language delay (71%), hypotonia (69%), epilepsy (63%), behavioural problems including autism spectrum disorders (47%) and brain abnormalities such as ventriculomegaly and a small corpus callosum (67%) [2]. Haploinsufficiency is the assumed disease mechanism leading to the clinical features. All reports so far have identified PPP2CA de novo mutations, including a partial gene deletion, a frame shift, three nonsense mutations, a single amino acid duplication, a recurrent mutation, and eight non-recurrent missense mutations [2].
The range of phenotypic characteristics of PPP2CA de novo mutation and its genetic heterogeneity are expanding due to the availability and increased use of genome sequencing in children with neurodevelopmental disorders.
We present a case of a novel de novo PPP2CA gene mutation (c.176A > G (p.H is 59 Arg)) and its associated neurological features. Both dystonia and progressive cerebral atrophy have not been included in any report published so far.
Case Presentation
A 3-year-old girl first presented to our Paediatric Neurology services at Chelsea and Westminster Hospital (CWH) at around 10 weeks of age due to poor weight gain (dropping from the 50th centile at birth to the 0.4th centile), gastro-oesophageal reflux, an unsafe swallow (requiring NG feeding) and global developmental delay, without language development.
She was born at 39 weeks of gestation by normal delivery in a good condition to non-consanguineous Kurdish parents. In terms of her development, at the age of 5 months, she had not achieved good head control, was not able to roll over, nor put her hands in her mouth, smile or fix and follow.
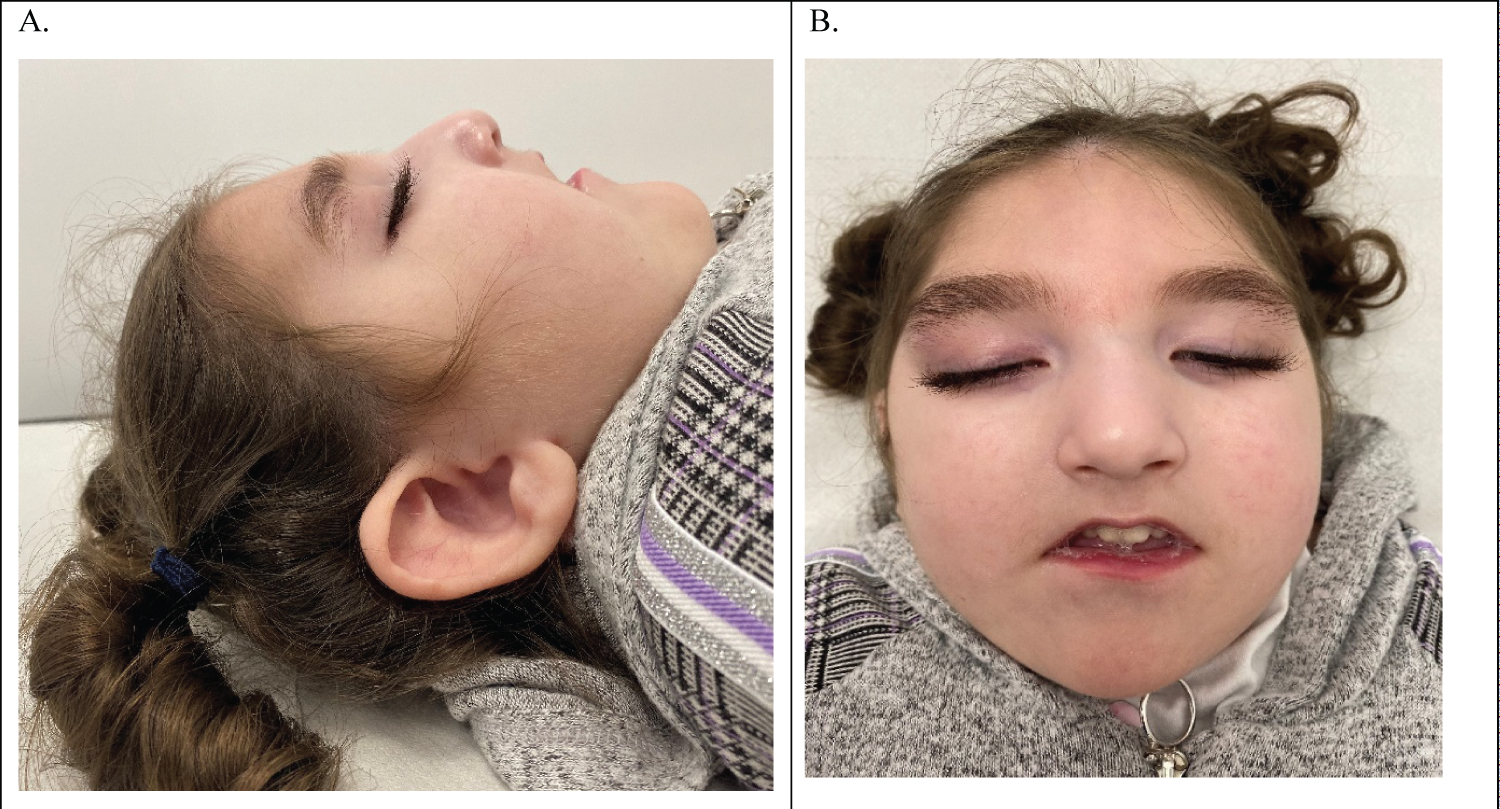
On her first examination, she was noted to be microcephalic with overlapping sutures (head circumference below the 0.4th centile), a small anterior fontanelle, high-arched palate, long eyelashes, down slanting and long palpebral fissures, subluxation of patellae, and hyper mobility of the thumb joints (Figure 1). She had increased tone in the upper and lower limbs with fisting of the hands and hyperreflexia and a tendency toward opisthotonic posture with dystonic movements.
Concerns about seizures began at around the age of 4 months, although in retrospect these episodes had probably started at around 2 months of age. These involved arms becoming flaccid with back arching and eyes rolling back for 2-4 seconds. She was initially started on levetiracetam (later built up to 60 mg/kg/day), however seizures evolved into infantile spasms with hypsarrhythmia detected on EEG at 11 months of age. She had up to 10 clusters of spasms in 5 minutes occurring 3-4 times a day. She was treated with vigabatrin and prednisolone with no success. Zonisamide was later added (building up to 10/kg/day). She was referred for a ketogenic diet therapy at this stage, but it was felt unsuitable for this candidate. DNA analysis using an Epileptic Encephalopathy gene panel gave normal results.
Since a very early age, she was noted with persistent dystonic episodes described as arching backwards (acquiring a 'banana-shape' posture), extending her upper limbs, pronating and fisting bilaterally, and extending her legs. She was started on trihexyphenidyl at around 5 months of age, which was quickly switched to clonidine (slowly built up to 1.38 mcg/kg/hr) due to prolonged exaggerated episodes of dystonia with increased secretions and desaturations. Later, gabapentin was also added (increasing to 67 mg/kg/day).
Ophthalmology and audiology reviews identified visual impairment with optic nerve hypoplasia and sensory-neuronal hearing impairment, respectively.
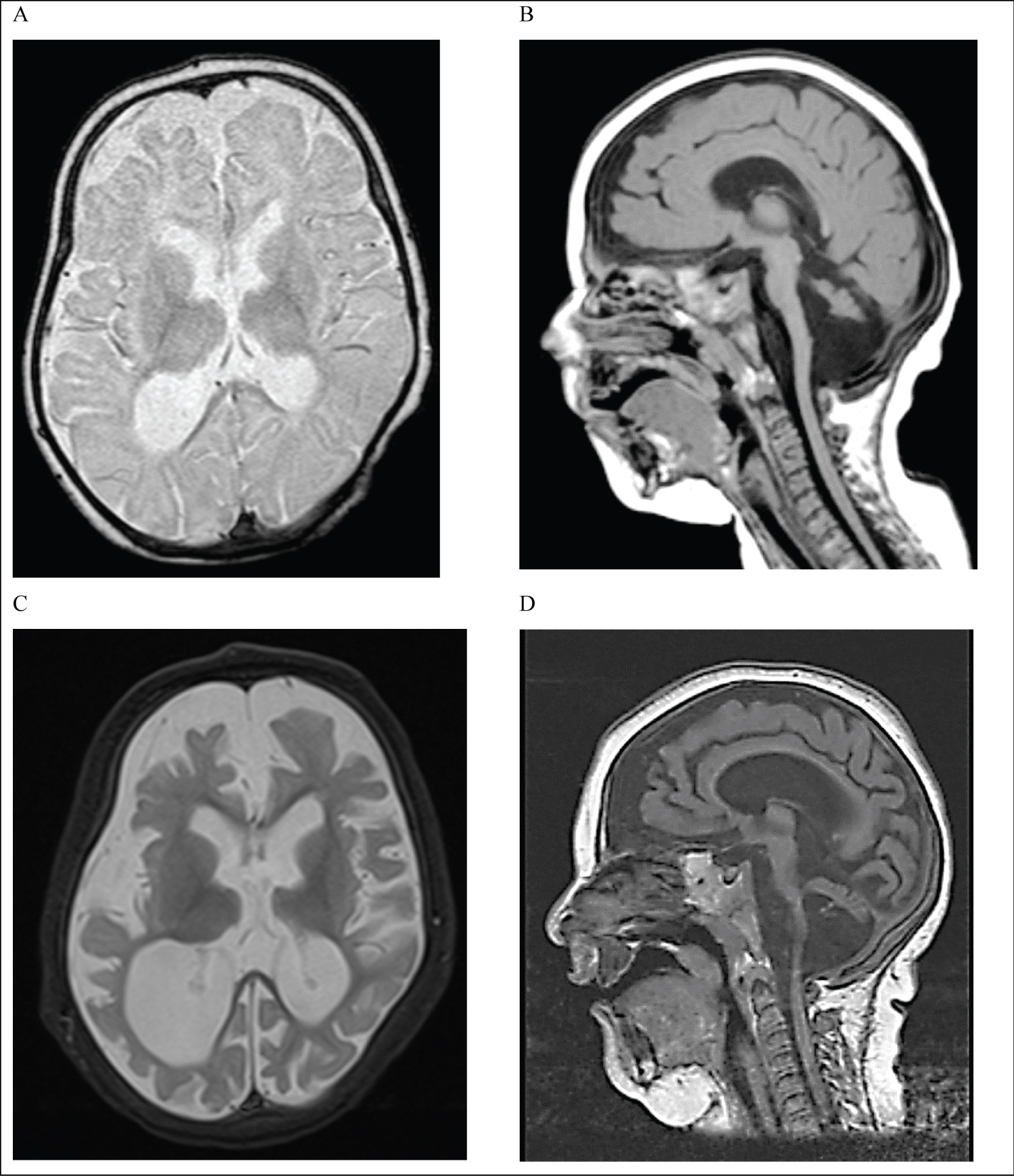
The patient had extensive investigation tests including neuro-metabolic screen (blood, urine and CSF tests), which was reported as normal. An MRI brain scan performed at 5 months of age showed a global underdevelopment of the brain. The findings were suggestive of an underlying neurometabolic or neurogenetic disorder (Figure 2). A repeat MRI scan carried out at 11 months (Figure 2) detected a progressive global brain atrophy, a marked pontocerebellar overlay, focal signal changes in brain stem nuclei and globally delayed myelination. At that time, DNA analysis using a gene panel for genes associated with pontocerebellar hypoplasia was reported normal.
A muscle biopsy was undertaken during anaesthetics for a gastrostomy insertion when the child was 14 months old. Analysis of muscle respiratory chain enzymes revealed loss of complex I and IV enzymatic activity. Based on these results and the latest MRI findings, an initial diagnosis of a probable mitochondrial disorder was suggested. The patient was started on Ubiquitine and Riboflavin. Subsequently, DNA analysis of the entire mitochondrial genome did not reveal any abnormality.
At the age of around 2½-years-old, as part of the 100K genome project, she was found to have de novo heterozygosity for the PPP2CA c.176A > G; (p.H is 59 Arg) variant on trio genome sequencing. The following gene panels were applied to the whole genome sequencing data: early onset dystonia, cerebellar hypoplasia, mitochondrial disorders, rare multisystem ciliopathy disorders, hereditary spastic paraplegia, undiagnosed metabolic disorders, congenital disorders of glycosylation, arthrogryposis, hereditary ataxia, genetic epilepsy syndromes, structural basal ganglia disorders, and intellectual disability.
Following an assessment by a metabolic team, it was believed that this was a secondary mitochondrial dysfunction rather than primary.
Her head circumference progressively further reduced. On the latest admission to the ward, we recorded reduction by 1 cm within the last 6 months. This was also accompanied by a clinical deterioration noted every 6 months and developmental regression.
Recently, her clinical status has significantly worsened with an increased seizure frequency, further reduction of her head circumference, further developmental decline, scoliosis and exaggerated dystonic features with Status Dystonicus (Stage IV). The latter required an admission to the paediatric high dependency unit. Later, her care was handed over to a palliative team, which prescribed further increase in the doses of her anti-epileptic (Zonizamide 12 mg/kg/day, Levetiracetam 60 mg/kg/day) and anti-dystonic medications (Clonidine 4 mcg/kg/day, Gabapentin 90 mg/kg/day, Chloral Hydrate 100 mg/kg/day), resulting in constant lethargy and sleeping.
Discussion
Our case report expands the phenotypical features associated with a PP2CA-related neurodevelopmental disorder to include dystonia and progressive brain atrophy.
The other features in our case fit well with the phenotypic presentation described in a recently published case series which reported 16 individuals with de novo mutations in PPP2CA [2]. Intellectual disability and developmental delay ranged from mild to profound in this group. There was also a large variation in the severity of associated morbidity wherein 7 individuals were diagnosed with autism spectrum disorders, 11 with hypotonia, 9 with feeding difficulties, 6 with vision problems, 3 with congenital heart defects and 10 with different types of seizures and epileptic syndromes. There was also a variation in MRI head scan findings with 4 having dilated ventricles, 2 delayed myelination, and 4 dysplasia or absence of the corpus callosum. Of these, 2 were macro cephalic and 4 were micro cephalic. Some were dysmorphic but without a specific pattern of facial features. Dystonia was not described in any of these cohorts of patients, nor mentioned in the progression of the cerebral atrophy on the brain MRIs. Furthermore, optic atrophy has not been described in this cohort, although, optic nerve anomaly was mentioned.
Pathogenic variants in PPP2CA are diverse and suggest loss of function as the pathogenetic mechanism [2]. The specific PPP2CA gene variant in our patient has not been reported before, although a pathogenic variant in the directly neighbouring amino acid (p.Gly60Val) has been reported in a 3-year-old boy, with only mild developmental delay, no seizures and a normal head size [2]. A clear genotype-phenotype relation is not apparent. The same wide clinical variability in PP2A protein dysfunction-related genes has been reported in PPP2RA1, correlating with the degree of biochemical dysfunction of the DNA variant in the gene [8].
The PPP2CA c.176A > G variant is absent from the Genome Aggregation Database (gnomAD) and multiple in silico tools predict it to be deleterious. PPP2CA has a low tolerance for missense variation. This variant is therefore considered likely to be pathogenic.
Progressive global cerebral atrophy detected on MRI scan is a pivotal feature in the clinical presentation of this case, causing progressive clinical decline of our patient's condition. Progressive cerebral atrophy and reduction of head circumference was not documented in the previously mentioned case series, however 2 individuals were noted to have neurodevelopmental regression, occurring after a psychotic episode in one of these cases [2]. Parents consistently denied consanguinity, thus making a second recessive genetic cause extremely unlikely. Features of severe brain atrophy are indeed surprising in this patient with neighbouring mutations not showing this phenotypical finding.
The second pivotal finding in our case report is a progressively deteriorating dystonic condition with presentation of status dystonicus (Stage IV). This feature was not reported in the 16 published cases in the literature, although one published patient was said to have another extrapyramidal movement disorder and choreatic movements [2]. This dystonia is not thought to be due to secondary causes, with MRI, metabolic and genetic panels ruling out differential causes for dystonia.
Interestingly, our patient had features of mitochondrial dysfunction noted on the muscle biopsy and respiratory chain enzyme investigations. Although this has not been extensively described before, in our clinical experience, abnormalities of mitochondrial complexes detected with muscle biopsy can happen in many genetic conditions as a non-specific feature, possibly secondary to metabolic changes associated with the underlying genetic defect. In clinical practice, these results can lead to a diagnosis of mitochondrial dysfunction and DNA analysis of mitochondrial genes, but in our opinion should not distract one from exploring an underlying genetic diagnosis if these turn out normal.
Conclusion
We present a 3-year-old girl with a novel DNA variant in the PPP2CA gene, namely c.176A > G (p.H is 59 Arg), associated with a novel clinical presentation of severe dystonic condition and progressive brain atrophy. These clinical features further expand the phenotype spectrum of this gene mutation, suggesting that PPP2CA gene mutations should be considered in the differential diagnosis of progressive microcephaly and paediatric neurodegenerative conditions.
References
- Janssens V, Goris J (2001) Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353: 417-439.
- Reynhout S, Jansen S, Haesen D, et al. (2019) De novo mutations affecting the catalytic Cα subunit of PP2A, PPP2CA, cause syndromic intellectual disability resembling other PP2A-related neurodevelopmental disorders. Am J Hum Genet 104: 139-156.
- de Ligt J, Willemsen MH, van Bon BW, et al. (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367: 1921-1929.
- Gilissen C, Hehir-Kwa JY, Thung DT, et al. (2014) Genome sequencing identifies major causes of severe intellectual disability. Nature 511: 344-347.
- Houge G, Haesen D, Vissers LE, et al. (2015) B56δ-related protein phosphatase 2A dysfunction identified in patients with intellectual disability. J Clin Invest 125: 3051-3062.
- Loveday C, Tatton-Brown K, Clarke M, et al. (2015) Mutations in the PP2A regulatory subunit B family genes PPP2R5B, PPP2R5C and PPP2R5D cause human overgrowth. Hum Mol Genet 24: 4775-4779.
- Shang L, Henderson LB, Cho MT, et al. (2016) De novo missense variants in PPP2R5D are associated with intellectual disability, macrocephaly, hypotonia, and autism. Neurogenetics 17: 43-49.
- Lenaerts L, Reynhout S, Verbinnen I, et al. (2021) The broad phenotypic spectrum of PPP2R1A-related neurodevelopmental disorders correlates with the degree of biochemical dysfunction. Genet Med 23: 352-362.
Corresponding Author
Dr. Naila Ismayilova, Consultant Paediatric Neurologist, Chelsea and Westminster Hospital, 369 Fulham Road, Chelsea, London, SW10 9NH, UK.
Copyright
© 2022 Cheng E, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.