CD59 Deficiency in Two Bahraini Children: Clinical Profile and Outcome
Abstract
CD59 is an inherited autosomal recessive disorder characterized by various clinical manifestations including hemolytic anemia, neurological deficit due to recurrent stroke and chronic inflammatory demyelinating polyneuropathy like picture. CD59 is a glycoprotein encoded by CD59 gene, and expressed in various body tissues. It provides protection against complement membrane attack complex. The aim of this presentation is to describe the clinical profile of a relative rare and unique condition, which has potential therapeutic approach and the diagnosis can help for genetic counseling.
Keywords
CD59 deficiency, Chronic inflammatory demyelinating polyneuropathy, CIDP, Eculizumab
Introduction
Hemolytic Anemia, CD59-Mediated, with or without Immune-Mediated Polyneuropathy (HACD59) (OMIM 612300) is an autosomal recessive disorder caused by mutation in CD59 gene on chromosome 11p13 [1]. CD59 is expressed on different cell lines, including hematopoietic cells, endothelial cells, neurons, and neuroglia, retinal pigment epithelial cells and it regulates the complement system by encoding a GPI-anchored cell surface membrane glycoprotein, that inhibit the formation of the membrane-attack complex (MAC) osmolytic pore formation [2,3]. The disease manifests early in infancy (3 to 7 months) with Prevalence of < 1/1000000 (Worldwide) [1]. The disease manifestations include hemolytic anemia in the form of Paroxysmal nocturnal hemoglobinuria (PNH), relapsing-remitting polyneuropathy exacerbated by infection, generalized hypotonia and hyporeflexia affecting the lower limbs more than the upper limbs, skeletal muscle atrophy, muscle weakness, acute promyelocytic leukemia, increased CSF protein, and macular degeneration [4].
The disease manifestation of PNH is associated with risk of thrombotic arterial events, presenting as strokes [5], and acute coronary syndromes [2]. The Complex of familial phenotype of early peripheral neuropathy, recurrent CNS ischemic like strokes and retinal involvement were considered previously to be due to mitochondrial disorders [4].
Case 1
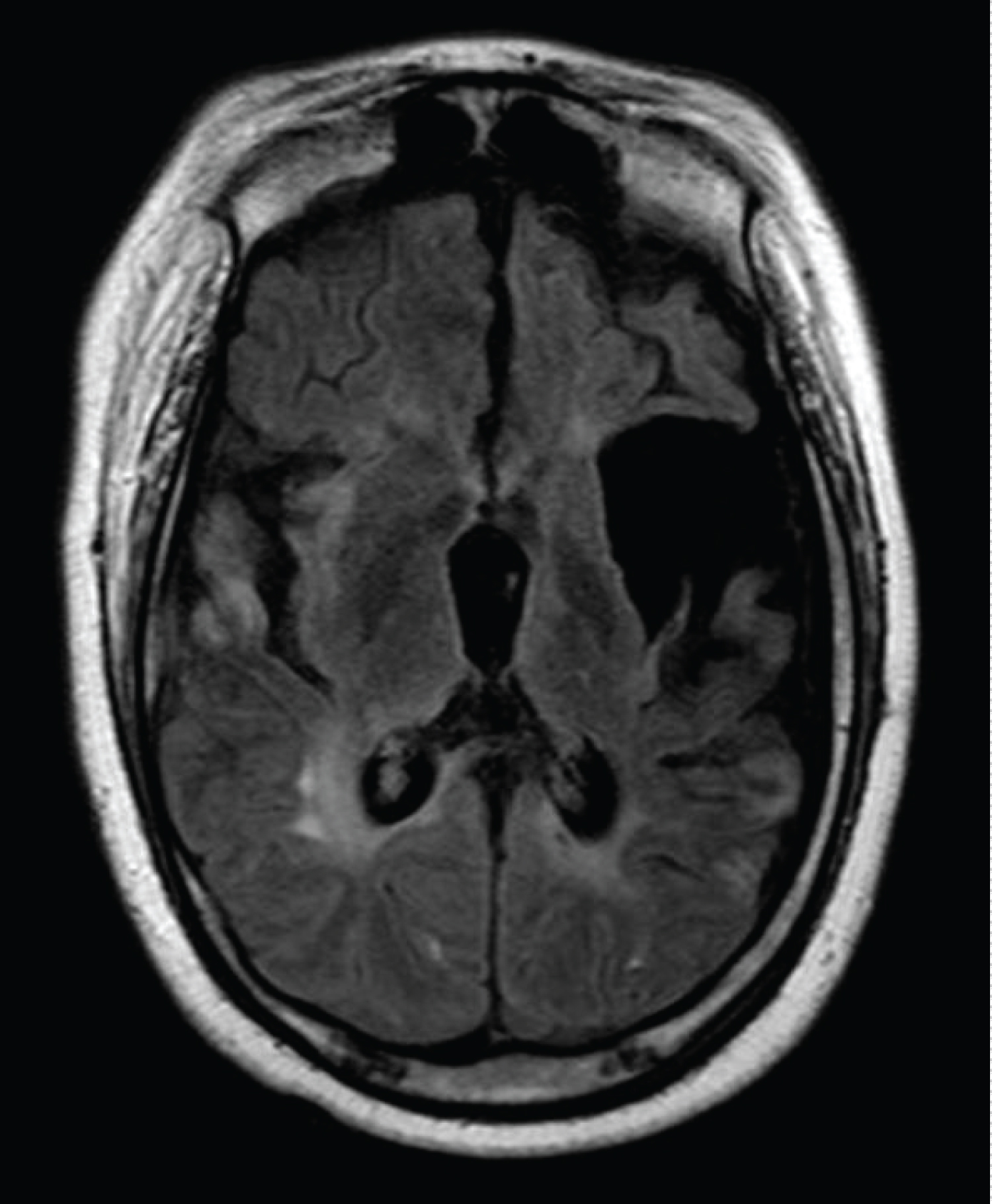
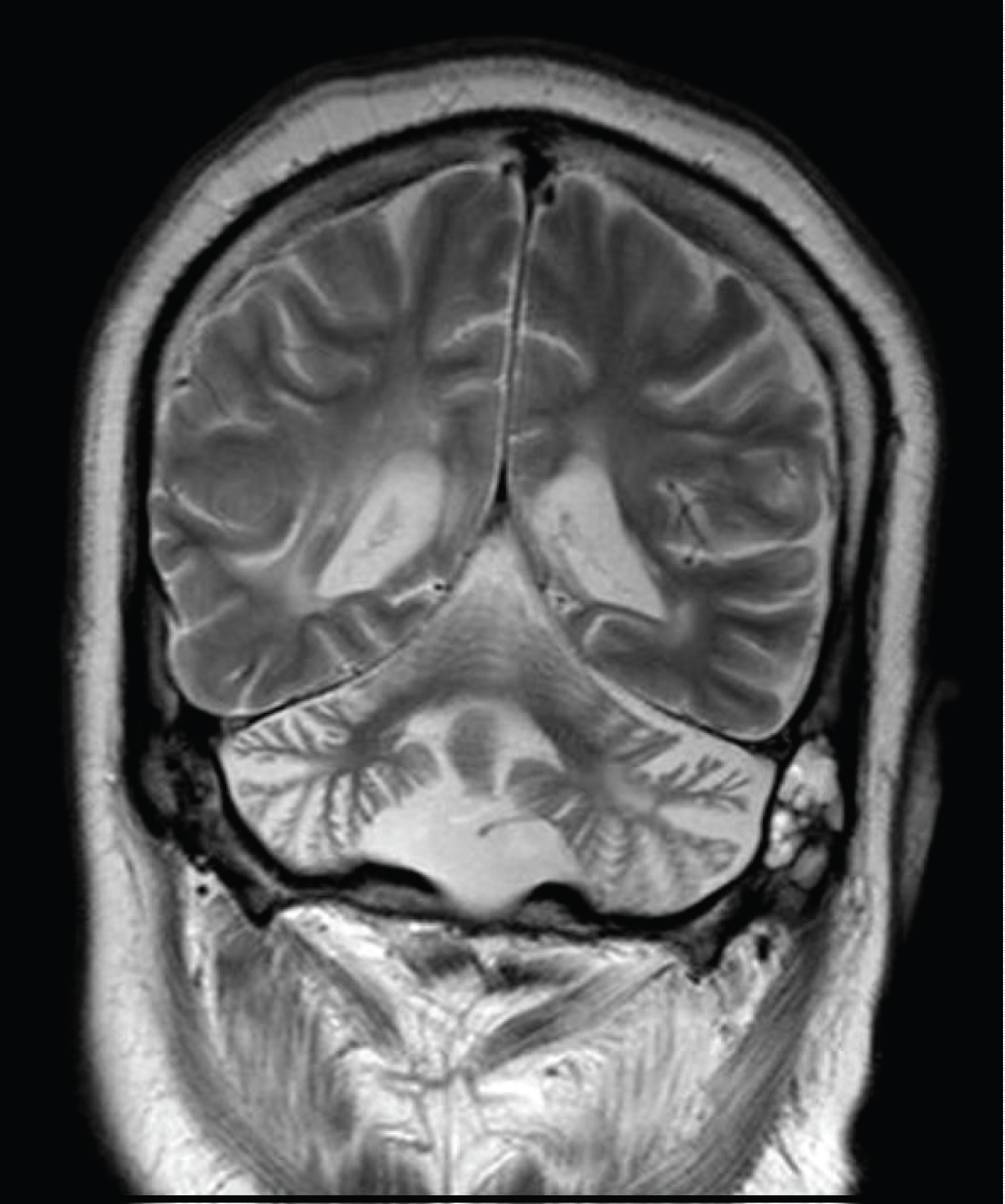
SM is a fifteen-year-old boy following in pediatric neurology clinic for many years. The boy was a product of full term, normal vaginal delivery with no perinatal complications; he achieved some developmental milestones initially. At the age of eleven months he was admitted with encephalopathy like picture and treated for seizure with antiepileptic medications. Later the boy gradually improved and eventually he was able to walk with left sided weakness, slurred speech and mental handicap; neuroimaging showed multiple regions of encephalomalacia, gliosis, demyelination and cortical/cerebellar atrophy, (see Figure 1 and Figure 2). He showed also features of peripheral neuropathy for which nerve conduction study revealed axonal neuropathy. Laboratory investigations for Tandem mass spectroscopy, organic acids, lactate, ammonia, prothrombotic disorders and vasculitis screen were unremarkable.
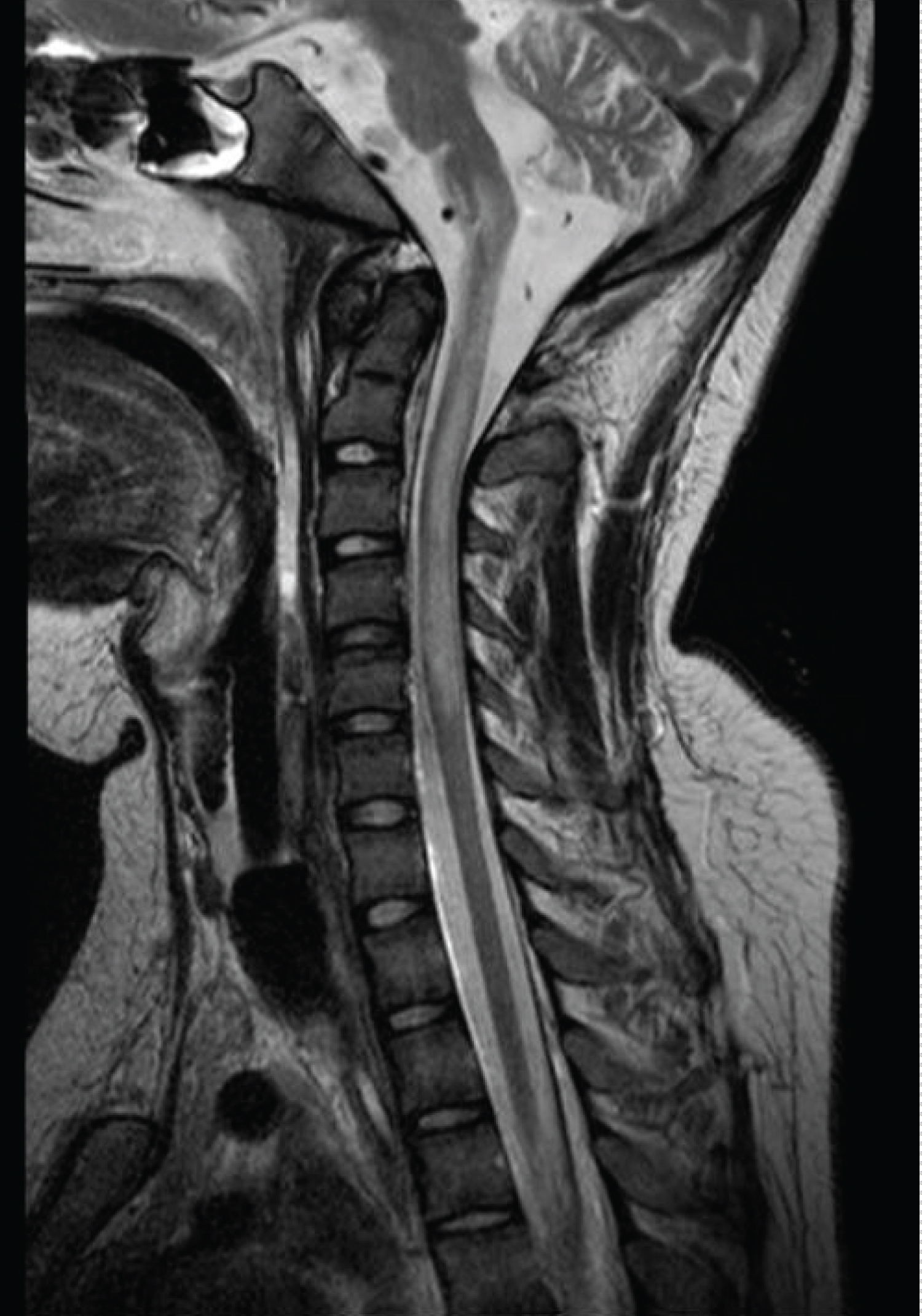
During his last admission, he presented with generalized body weakness and facial angioedema. He was lethargic, opening eyes, fixing, with poor verbal communication, left sided facial weakness, poor gag reflex; generalized body weakness with flaccid paralysis involving all extremities with absent deep tendon reflexes, sensation could not be assessed. He required intubation and ventilation. MRI of the spine showed extensive cervical cord lesion from C3-C7 with high intensity but without enhancement; no new brain lesion was seen, (see Figure 3). The boy had recurrent strokes previously, peripheral neuropathy but this time he has picture of acute transverse myelitis. He was managed with steroids and plasma exchange sessions, but unfortunately he remained ventilator dependent and died from sepsis.
Case 2
EM is a six-year-old girl; a product of full term normal delivery with birth weight of 3 kilograms. By reviewing history the condition started at the age of 20 months of age as the parents reported three events of generalized body weakness at times with facial weakness with inability to stand or walk provoked by febrile illness. Case 1 is her elder brother.
On her first examination, the girl was awake and attentive, extra ocular movements were full, pupils were equal and reactive to light, no facial symmetry, and the tongue in the midline no deviation or fasciculation, saying few words, but clumsy. Motor exam showed mild hypotonia with waddling gait, and absent deep tendon reflexes. 2 years Later the girl presented with sudden onset partial visual loss, she was not able to recognize colors, she can see waving hands and was able to walk independently without hitting objects. Ophthalmological evaluation showed retinal dystrophy with suspicious retinitis pigmentosa. Her initial MRI brain was unremarkable. Nerve conduction study showed motor neuropathy of demyelinating type. The girl's vision recovered later.
In view of these features the girl was suspected to have mitochondrial disorder and she was started accordingly on L-Carnitine and Coenzyme Q10. A trial of IVIG was planned to be given but the parents declined for social reasons.
One year later the girl presented with acute encephalopathy and recurrent seizure which required intubation, she was managed with antibiotics, Methyl Prednisolone pulse therapy & Acyclovir. Later on examination she had spastic quadriparesis and bulbar weakness. Her MRI brain showed extensive high signal intensity involving the thalami, basal ganglia, brain stem and occipital lesion bilateral. She had evidence of Epilepsia Partialis Continua and she remained on Levetiracetam and Topiramate.
She was extubated and showed minimal recovery at home, and remained bedridden.
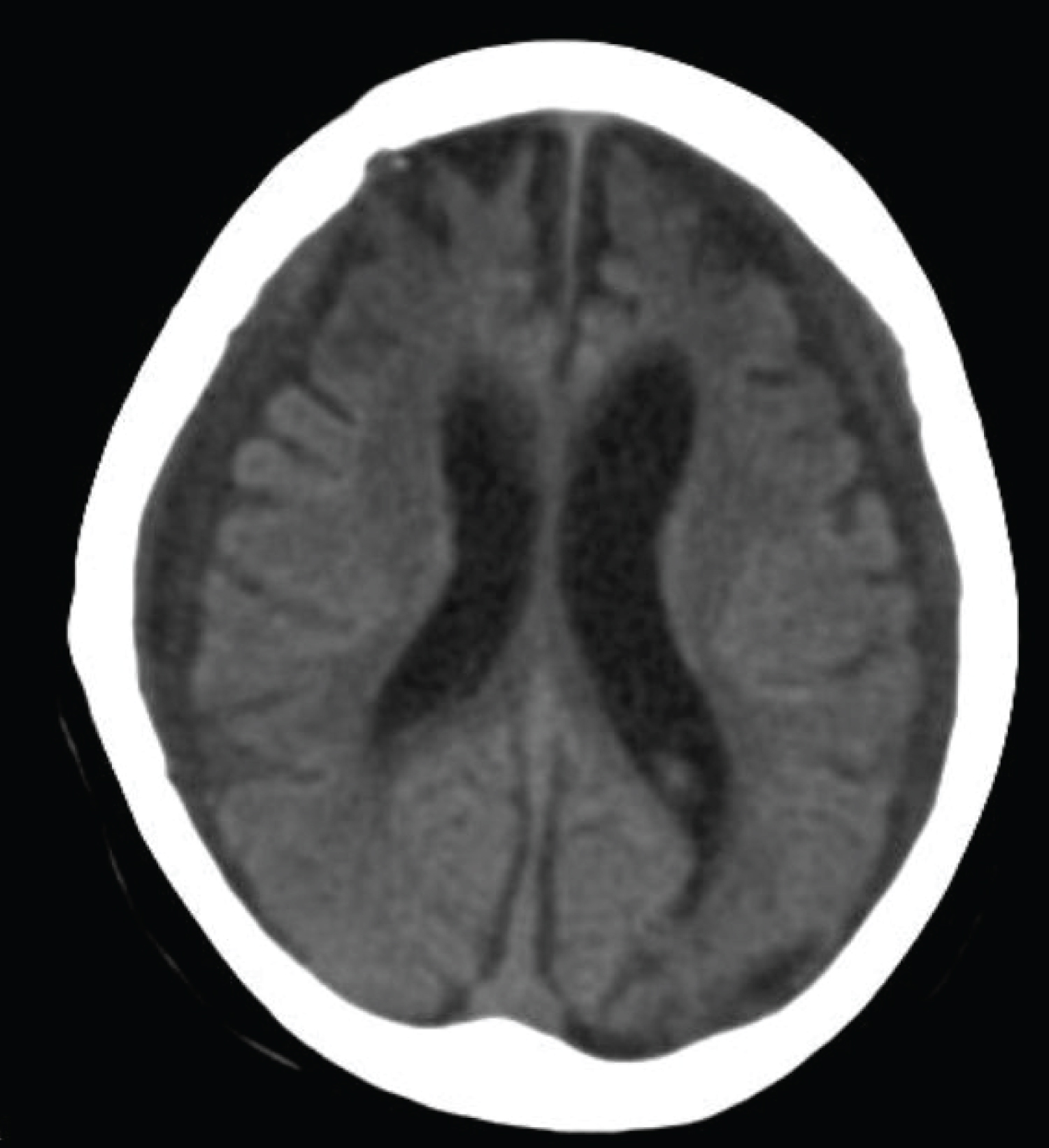
One year later the girl presented with shallow breathing and seizure, she required immediate intubation for poor respiratory effort. The girl had further deterioration in her sensorium and went into stupor state, there was unequal pupils toward the left pupil. CT brain showed ventricular dilatation and brain atrophy, external ventricular drain inserted which was replaced later by ventriculoperitoneal shunt, (see Figure 4). The girl was in a poor neurological condition with features of minimal conscious state, subsequently she died from sepsis.
The diagnosis was confirmed by molecular DNA sequencing of CD59 gene which revealed a homozygous; c.323C>A, p.Ser108*. Its associated with hemolytic anemia, CD59 mediated, with or without polyneuropathy. Parents were carrying the mutation in heterozygous state.
Discussion
Neurogenetic disorders are heterogeneous groups of neurological conditions caused by defective genes. They may affect different parts of the nervous system either central or peripheral or both. They can be associated with extra neural manifestations as well. Thousands of neurogenetic conditions have been described so far. The journey of thousand miles begins with one step, hence reaching the diagnosis of a rare disease is a cumbersome task. Begins with collecting clinical data, meticulous physical examination, reviewing the various diagnostic tests including neurophysiological and neuroimaging. Eventually, you may characterize the phenotype of the condition. The advent in genetic testing including whole exome sequencing opened venue for better diagnosis and understanding the nature of the disease. Exome sequencing is a genomic technique for sequencing all of the protein coding regions in a genome consisting about 1% of the human gene [6].
Exome sequencing can identify genetic mutation causing neurological disease which can have major implications for therapeutic approaches and testing at risk family members [7]. CD59 deficiency is frequently seen in patients with paroxysmal nocturnal hemoglobinuria (PNH).
CD59 is a glycoprotein that is linked to cell membranes by glycosyl-phosphatidyl inositol (GPI) anchor, is widely distributed, being expressed not only on peripheral blood cells but also on cells in various organs [8]. CD59 binds to both C8 and C9 in assembling membrane attack complex (MAC) to provide protection from complement mediated lysis [9]. In a study by Vedeler, et al. they found CD59 on cells in situ and in culture from normal nervous tissue as well as soluble in the cerebrospinal fluid (CSF). CD59 was present on schwann cells, neurons and endothelial cells in the peripheral nervous system. In the central nervous system CD59 was found predominantly on endothelial cells. There was also diffuse staining of white and gray matter of the spinal cord and the brain, presumably of microglia, oligodendrocytes, astrocytes and neurons as these cells were CD59 positive in culture [10]. CD59 deficiency is a common finding in adult patients with paroxysmal nocturnal hemoglobinuria (PNH). Chronic inflammatory demyelinating polyneuropathy (CIDP) is an immune-mediated disorder characterized by progressive or relapsing motor and sensory deficits. The estimated prevalence in children is 0.48/100000 [11]. Pediatric CIDP is generally a chronic condition, often requiring multiple modalities of treatment over time after leaving children with residual deficits even with successful treatment, a majority of patients do not have complete remission of their illness, and many need intermittent if not continuous immunomodulating treatment [12]. Haliloglu, et al. described 3 affected family members with early onset immune mediated axonal neuropathy, cerebrovascular event both in the anterior and posterior circulation, and chronic coombs-negative hemolysis, due to CD59 deficiency diagnosed through genetic testing and flowcytometry [5]. One girl died at the age of 16 due to pneumonia, second child was managed with serial IVIg courses, she was still ambulant but with some difficulties in gait. The third boy was started eventually on Eculizumab. Another case series described by Nevo, et al. consisting of five children of north africanjewish origin from four unrelated families a homozygous missense mutation p.Cys89Tyr in CD59 was identified in all patients, which is associated with a failure of proper localization of the CD59 protein in the cell surface [12]. Recently Solman, et al. described a unique condition with recurrent episode of demyelination in the central nervous system as the only manifestation of inherited CD59 deficiency, the patient had partial response to IVIG and steroid treatment. The unfortunate siblings were both affected profoundly the boy had recurrent encephalopathy with stroke like events with partial recovery (Table 1). As mentioned earlier, extensive investigations including metabolic screen did not reveal the etiology. The boy developed myelopathy as well which made him quadriplegic and remained ventilator dependent, the boy had episodes of hematuria during his admission which was not explained as well as his facial angioedema. While his sister hematuria was explained by renal calculi. Overall, their anemia was subtle, there was no gross hemolysis. The introduction of whole exome sequencing was quiet helpful to elucidate the etiology. Both kids were treated with standard medications including IVIg and steroids and they showed partial benefit.
Although Eculizumab said to have a role in the treatment in children with CD59 mutation; their condition by the time of diagnosis was in poor situation, the girl was already in persistent vegetative state and the boy was quadriplegic for long time and suffered from recurrent infections, sepsis and pneumonia, so Eculizumab was not offered. CD59 is expressed in neurons; low neuronal expression results in neural susceptibility to MAC-driven lysis [13].
Conclusion
CD 59 deficiency need to be considered in the differential diagnosis in a child with recurrent relapses of neuropathy, stroke and hemolytic anemia; immunomodulatory treatment need to commenced to mitigate the deficits of the disease in addition for family counselling purpose.
References
- https://omim.org/entry/612300.
- Tabib A, Karbian N, Mevorach D (2017) Demyelination, strokes, and eculizumab: Lessons from the congenital CD59 gene mutations. Mol Immunol 89: 69-72.
- Höchsmann B, Dohna-Schwake C, Kyrieleis HA, et al. (2014) Targeted therapy with eculizumab for inherited CD59 deficiency. N Engl J Med 370: 90-92.
- Ben-Zeev B, Tabib A, Nissenkorn A, et al. (2015) Devastating recurrent brain ischemic infarctions and retinal disease in pediatric patients with CD59 deficiency. European Journal of Paediatric Neurology 19: 688-693.
- Haliloglu G, Maluenda J, Sayinbatur B, et al. (2015) Early-onset chronic axonal neuropathy, strokes, and hemolysis: inherited CD59 deficiency. Neurology 84: 1220-1224.
- Ng SB, Turner EH, Robertson PD, et al. (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461: 272-276.
- Ng SB, Buckingham KJ, Lee C, et al. (2010) Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42: 30.
- Vedeler C, Ulvestad E, Bjørge L, et al. (1994) The expression of CD59 in normal human nervous tissue. Immunology 82: 542.
- Meri S, Waldmann H, Lachmann PJ (1991) Distribution of protectin (CD59), a complement membrane attack inhibitor, in normal human tissues. Lab Invest 65: 532-537.
- Davies A, Simmons DL, Hale G, et al. (1989) CD59, an LY-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. The Journal of Experimental Medicine 170: 637-654.
- Desai J, Ramos-Platt L, Mitchell WG (2015) Treatment of pediatric chronic inflammatory demyelinating polyneuropathy: Challenges, controversies and questions. Annals of Indian Academy of Neurology 18: 327-330.
- Nevo Y, Ben-Zeev B, Tabib A, et al. (2013) CD59 deficiency is associated with chronic hemolysis and childhood relapsing immune-mediated polyneuropathy. Blood 121: 129-135.
- Yamashina M, Ueda E, Kinoshita T, et al. (1990) Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. N Engl J Med 323: 1184-1189.
- Motoyama N, Okada N, Yamashina M, et al. (1992) Paroxysmal nocturnal hemoglobinuria due to hereditary nucleotide deletion in the HRF20 (CD59) gene. Eur J Immunol 22: 2669-2673.
Corresponding Author
Ayman Khalil Ali, Department of Neuroscience, Salmaniya Medical Complex, Ministry of Health, Kingdom of Bahrain.
Copyright
© 2021 Ali AK, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.