Intracranial and Ocular Abnormalities in a Child with Neurodevelopmental Delay
Abstract
Background: COL4A1 mutations can mimic TORCH infections and should be considered in the differential of congenital infections, especially when additional neuroanatomical abnormalities exist.
Case presentation: A patient with neurodevelopmental delay and an unremarkable prenatal and birth history presented postnatally with congenital cataracts and neuroanatomical abnormalities including periventricular calcifications, porencephaly, and cerebellar hypoplasia. Although there was initial suspicion for a TORCH infection including cytomegalovirus, further genetic testing revealed a novel COL4A1 mutation, which involves type 4 collagen alpha 1 chain, an important component of vasculature.
Conclusions: This case highlights the neuroanatomical and extra cranial features of COL4A1 mutation which helps differentiate the condition from other related diseases. This report suggests that COL4A1 should be considered in a child with intracranial and ocular abnormalities, particularly in the absence of a perinatal etiology.
Keywords
COL4A1, Intracranial calcification, Porencephaly, Cataract, Developmental delay, TORCH
Abbreviations
COL4A1: Type IV Collagen; HANAC: Hereditary Angiopathy with Nephropathy Aneurysm and Muscle Cramps; TORCH: Toxoplasmosis Rubella Cytomegalovirus Herpes Simplex Virus Parvovirus B19; CMV: Cytomegalovirus; PVL: Periventricular Leukomalacia
Introduction
COL4A1 mutations involve the type 4 collagen alpha 1 chain, which is an integral component of vascular basement membranes, particularly in the brain, eyes and kidneys [1]. Neurologically, the COL4A1 can result in a wide spectrum of abnormalities. The resulting fragility of the vasculature predisposes patients to prenatal and postnatal cerebrovascular events including micro and macrohemorrhages and ischemic damage resulting in white matter disease, calcifications and periventricular leukomalacia [2]. Porencephaly, or fluid-filled cavities in the brain, develop as a result of the intrauterine or neonatal hemorrhage [3], which manifests commonly as severe developmental delay, hemiplegia, and refractory seizures [4]. Silent microhemorrhages occur most commonly in the basal ganglia, supratentorial white matter, and cerebellum. COL4A1 can also result in defects in cortical development, including schizencephaly, polymicrogyria, and focal cortical dysplasia [4]. Cerebellar hypoplasia and other posterior fossa abnormalities can present in COL4A1, but are less common manifestations of the disease [1].
COL4A1 can also affect other organ systems [5]. Ocular abnormalities include congenital cataracts, which is the most commonly described ophthalmologic phenotype. Patients may also show retinal tortuosity, retinal hemorrhages, anterior chamber abnormalities, bilateral micro cornea, and unilateral retinal detachment. Renal manifestations present as hematuria, bilateral cysts of the kidneys and hereditary angiopathy with neuropathy, aneurysm, and muscle cramps (HANAC) syndrome [6]. Related musculoskeletal conditions include Walker Warburg syndrome, which is defined by ocular abnormalities, neuronal migration disorders, and myopathy.
Given the many associated clinical features affecting multiple organ systems, it is important to make the diagnosis of COL4A1 [2] early on to allow for appropriate investigation and management. Here, we present a case of a patient with neurodevelopmental delay who presented with neuroimaging findings and extracranial manifestations. This case informs an approach to differentiating COL4A1 from other similar presentations.
Case Presentation
A 4-year-old girl presented with profound developmental delay, cortical blindness, and epilepsy. She was born to non-consanguinous parents at 40 weeks and 2 days with an unremarkable prenatal history. She did not require resuscitation or a NICU stay. There was no contributory family history. Postnatally, she was found to have mild jaundice and congenital cataracts. She subsequently developed refractory seizures requiring multiple antiepileptic medications.
On examination, her head circumference was two standard deviations below normal. She had roving nystagmus and was unable to fix and follow. She had hypertonicity of her right upper extremity, limiting her ability to do fine motor tasks.
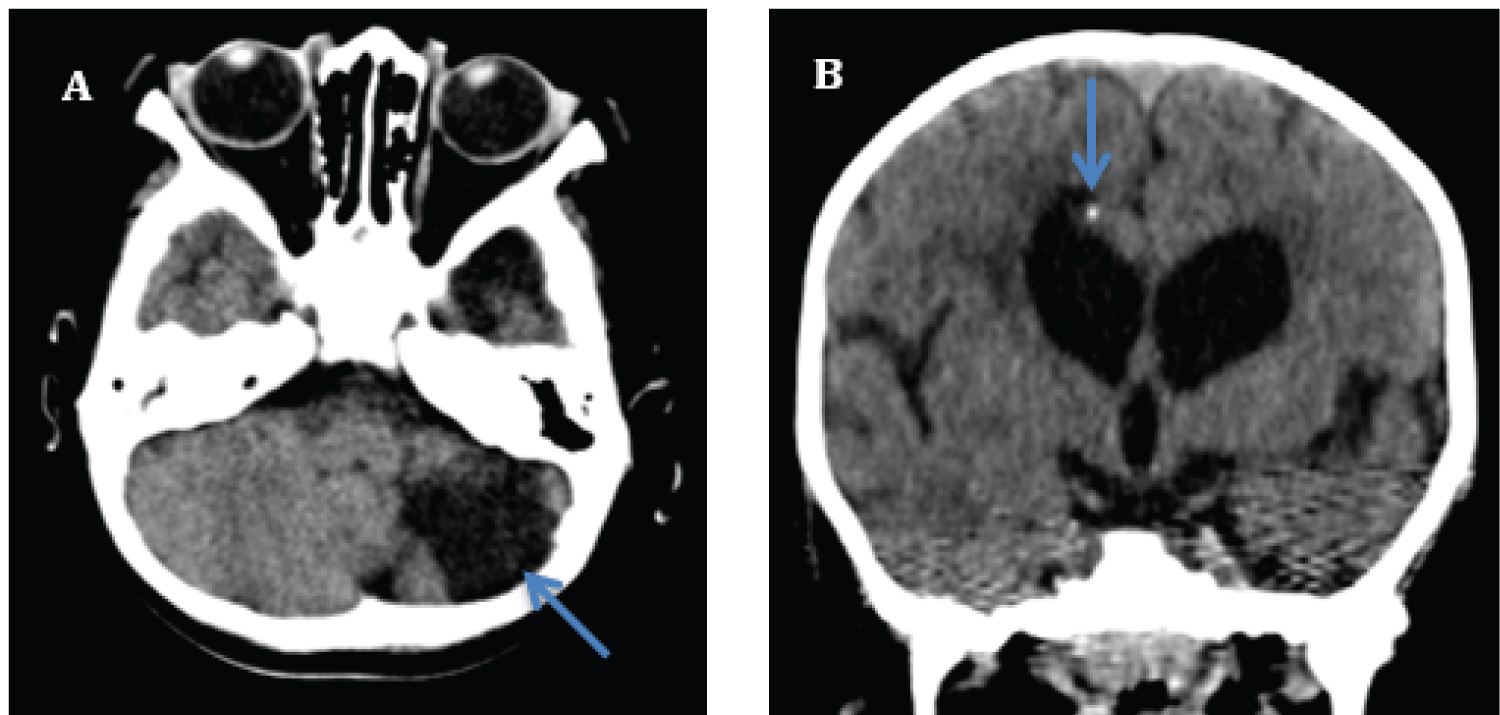
Imaging showed periventricular calcification, left cerebellar hypoplasia, dilatations of the lateral ventricles with porencephaly and white matter volume loss (Figure 1). Significant investigations were conducted including a chromosomal microarray, metabolic testing, genetic testing for Rett syndrome and Aicardi-Goutières syndrome. She was also reviewed by the Infectious Diseases team and a TORCH infection was ruled out. Thus, the patient and her parents were enrolled in a further whole exome sequencing study. The patient was ultimately found to have a de novo COL4A1 mutation.
Discussion
This case describes a patient with neurodevelopmental disorder of unknown cause. The patient demonstrated right spastic hemiplegia and neurological abnormalities arising early in development. In addition, there was evidence of extracranial comorbidities including congenital cataracts and roving nystagmus.
Consistent with the reported cases [2], this patient presentation included significant developmental delay, refractory epilepsy, and neuroanatomical abnormalities that could not be attributed to any perinatal etiology. There was also ocular involvement, in line with previous reports, including visual deficits, cortical blindness, and cataracts. Neuroanatomically, findings of porencephaly and intracranial calcifications were common. In this case, however, the patient also showed cerebellar hypoplasia, which is a rare feature but may provide additional evidence of COL4A1.
The patient's condition may have reflected many underlying etiologies. The precise locations of the calcification in the periventricular area suggested an early pathological process during which neurogenesis and neuronal migration occur. The differential included infectious causes such as CMV, Aicardi-Goutières syndrome, or PVL secondary to complications of prematurity or other acquired perinatal causes. Investigations for all these etiologies were negative in the current case.
Although periventricular calcifications are shared by many conditions, the porencephaly and cerebellar hypoplasia observed in this case are not seen in CMV, Aicardi Goutières Syndrome, and PVL arising from prematurity [2]. Moreover, the reported patient presented with ocular abnormalities, which suggests a more systemic vascular disease. Investigations into renal and musculoskeletal abnormalities were not done but may have added additional information to confirm the diagnosis.
COL4A1 can mimic multiple conditions including TORCH infections and ACS. However, unique brain abnormalities can be used to differentiate COL4A1 from other related diseases. Moreover, the presence of extracranial manifestations of disease including ocular abnormalities, nephropathy, and muscle cramps should prompt investigation into systemic involvement of COL4A1 mutation. These unique clinical features can help clinicians identify when to investigate for COL4A1 in a child with neurodevelopmental delay.
Declarations
Acknowledgements
Not applicable.
Funding
This research was supported by the Children's Hospital of Eastern Ontario. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Consent for publication
Written consent was obtained from the patient for publication of this case report and any accompanying images.
Competing interests
The authors declare no conflicts of interest with respect to the research, authorship, funding, and/or publication of this article.
Conflicts of interest
We have no conflicts of interest.
References
- Vehedi K, Alamowitch S (2011) Clinical spectrum of type IV collagen (COL4A1) mutations: A novel genetic multisystem disease. Curr Opin Neurol 24: 63-68.
- Livingston J, Doherty D, Orcesi S, et al. (2011) COL4A1 mutations associated with a characteristic pattern of intracranial calcifications. Neuropediatrics 42: 227-233.
- Yoneda Y, Haginoya K, Arai H, et al. (2012) De novo and inherited mutations in COL4A2, encoding the type IV collagen alpha2 chain cause porencephaly. Am J Hum Genet 90: 86-90.
- Zagaglia SZ, Selch C, Nisevic JR, et al. (2018) Neurological phenotypes associated with COL4A1/2 mutations. Neurology 91: 2078-2088.
- Smigiel R, Cabala M, Jakubiak A, et al. (2016) Novel COL4A1 mutation in an infant with severe dysmorphic syndrome with schizencephaly, periventricular calcifications, and cataract resembling congenital infection. Birth Defects Res A Clin Mol Teratol 106: 304-307.
- Alamowitch S, Plaisier E, Favrole P, et al. (2009) Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology 73: 1873-1882.
Corresponding Author
Dr. Meghna Rajaprakash, MSc, MD, Department of Neurology and Developmental Medicine, Kennedy Krieger Institute/Johns Hopkins Hospital, 716 N. Broadway, Baltimore, MD, 21205, USA
Copyright
© 2021 Rajaprakash M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.