Antenatal Counselling in Developmental Delay and Intellectual Disability: Case Series
Keywords
Antenatal counselling, Developmental delay, Intellectual disability, Cerebral creatine deficiency, X linked mental retardation, Hereditary spastic paraplegia
Introduction
Intellectual disability and developmental delay are common presentations encountered in pediatric clinical practice. The etiologies of these illnesses are diverse. Subtle clinical markers may act as pointers to underlying genetic etiologies in some of these conditions. Due to the increased availability of genetic tests, there is a robust role of genetic counselling in many of these conditions. However, investigations need to be judiciously advised in order to balance the anticipated gains with the expenses, more so in resource-limited settings. With this background, authors wish to report a series of cases where children had presented with various spectra of developmental delay, wherein, after clinical examination and appropriate testing, genetic diagnoses were unearthed and antenatal genetic counseling was offered.
Cases 1, 2
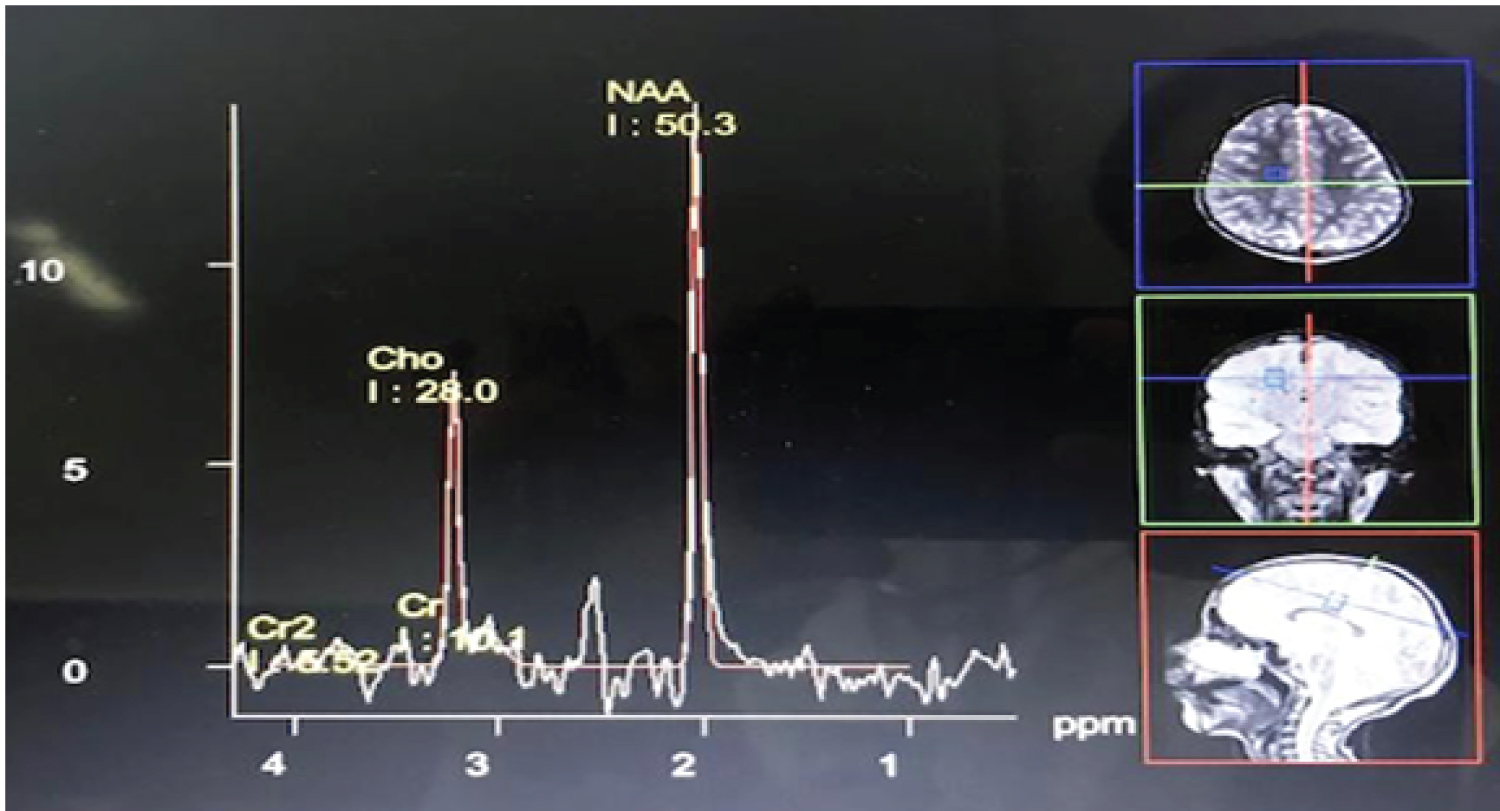
Two female siblings aged 7-years and 9-years, born to non-consanguineous parents were being followed up for intellectual disability and behavioural problems. Both children were born at term with uneventful antenatal/perinatal and post-natal periods. Their milestones were delayed. Their best milestones included ability to hop, feed self, speak short phrases and identify 6-7 body parts. They could not indicate bowel or bladder needs. They had certain behavioral peculiarities in the form of self-absorbed behavior, intermittent episodes of unprovoked laughter and crying spells and temper tantrums. The elder sibling had been having episodes of generalized tonic-clonic seizures lasting for 3-5 minutes per episode, which began at about 3-years of age occurring approximately once every six months. The child was seizure-free for nine months prior to presentation at the hospital (on oral, sodium valproate @ 30 mg/kg/day). There was no history of loss of developmental milestones, visual/hearing impairment and abnormal smelling urine/sweat. On examination, both children had poor eye contact and mood lability with laughter spells. The head circumferences of the elder and younger sibling were 44 cm and 46.5 cm respectively (both values < -3z WHO reference). Parental head circumferences were within normal limits. There were no neurocutaneous markers/dysmorphism. Their Intelligence Quotients (I.Q.) were 42 and 35 respectively. Examination of cranial nerves, motor, sensory, cerebellar and autonomic systems were unremarkable. Children were evaluated two years earlier. The elder sibling's karyotype and MRI brain were normal. Both children's metabolic screens and genetic testing for Angelman Syndrome were negative. Their mother presented at 6 weeks period of gestation (POG) for periconceptional testing. Hence, the children's diagnosis was revisited. In view of the probable familial nature of disease, underlying genetic etiology for intellectual disability (ID) with behavioral problems was suspected. Magnetic Resonance Imaging (MRI) brain with Magnetic Resonance Spectroscopy (MRS) of the elder sibling was done which revealed decrease in creatine peak (Figure 1). Her cerebrospinal fluid (CSF creatine levels were decreased. Her clinical exome sequencing revealed an autosomal recessive homozygous mutation in GAMT gene on exon 5 (Table 1) confirming the diagnosis of Cerebral Creatine Deficiency Syndrome-2. Chorionic villus sampling (CVS) was done, which revealed the GAMT mutation in heterozygous state (Table 1). Hence pregnancy was continued with an uneventful delivery. The infant is healthy and on follow-up. The siblings are being managed with protein-restricted diet, occupational therapy and special schooling. The eldest child is seizure free on oral levetiracetam therapy which was initiated as a replacement for sodium valproate. Creatine monohydrate supplementation, a recognized therapy for this condition,could not be exhibited due to availability issues. Both children are gaining milestones with improvement of behaviour and reduction of temper tantrums.
Case 3
5-year-old boy, born to non-consanguineous couple at term, was being followed up in pediatric OPD as a case of developmental delay. The child could climb up and down stairs but could not hop. He could scribble but could not feed self, babble but could not speak words and inhibited to 'no' but did not have stranger anxiety. He used to have seizures in the form of tightening of all limbs and uprolling of eyes, lasting for 5-10 minutes per episode from the age of 1-year, with frequency of 2-3 month on two antiepileptic drugs. He used to have self mutilating behaviour in the form of biting his fingers and knuckles compulsively, injuring himself on several occasions. There was no history of developmental regression or abnormal smelling urine/sweat. On examination, the child did not have dysmorphism/neurocutaneous markers. His anthropometry was unremarkable. Neurological examination revealed poor interaction with examiner and surroundings, normal motor/sensory/cerebellar and autonomic examination. Investigations revealed normal skeletal survey, thyroid function test, arterial ammonia, homocysteine, Arterial Blood Gas (ABG), Tandem Mass Spectrometry (TMS) and urinary Gas Chromatography Mass Spectrometry (GCMS). The child's mother presented at six weeks POG for periconceptional counseling. Hence clinical exome sequencing of the child was performed which yielded a known X-linked mutation of SYP gene in hemizygous state confirming the diagnosis of X Linked Mental Retardation Type 96 (Table 2). The mother's genetic testing revealed the same mutation in heterozygous state. Her amniotic fluid Deoxyribonucleic Acid (DNA) revealed the aforementioned mutation in hemizygous state (Table 2). Subsequently, medical termination of pregnancy was performed with parental consent.
Case 4
6-year-old boy born to 3° consanguineous parents with uneventful prenatal/perinatal periods was under pediatric follow-up as a case of cerebral palsy. He had intellectual disability and walking difficulty. He did not have any developmental regression, seizures, marked behavioral problems. There was no history of neurological illness in the family. His general examination was within normal limits. Examination of his higher mental functions revealed poor interaction and inability to follow one-step command. He had spasticity of bilateral lower limbs, decreased power of anti-gravity muscles (Medical Research Council grade 3/5), brisk deep tendon jerks and extensor plantars. He had contractures of bilateral tendo-achilles tendons. The child had a paraparetic gait. Examination of the other systems, skull and spine were within normal limits. Rest of his neurological examination was unremarkable.
His MRI brain revealed thinning of corpus callosum with delayed myelination. His parents had presented for antenatal counseling. In view of history of consanguinity and absence of perinatal risk factor, cerebral palsy seemed an unlikely diagnosis. His TMS and GCMS were normal and clinical. Clinical exome sequencing revealed homozygous mutation of AP4B1 gene diagnostic for spastic paraplegia type 47 (Table 3). Chorionic villus sampling (CVS) genome analysis revealed the above mutation in heterozygous state in both parents and in homozygous state in the fetus (Table 3). Hence, termination of pregnancy was done after parental consent.
Discussion
Intellectual disability is a relatively common condition with a prevalence of approximately 2.5% in the population [1]. Global developmental delay and intellectual disability may have an underlying genetic etiology in approximately 40% cases [2].
Cerebral Creatine Deficiency is a rare inborn error of metabolism involving creatine pathway comprising of three different disorders, among which guanidinoacetate methyltransferase deficiency (GAMT; MIM 601240) is the rarest variant which cause the autosomal recessive condition Cerebral Creatine Deficiency Type 2 [3]. About 150 cases of Cerebral Creatine Deficiency Type 2 have been reported till date [4]. Clinical manifestations of this entity include severe intellectual disability, autism, speech delay, seizures and extra pyramidal movements. MRS classically reveals decreased creatine peak [5]. Serum creatine and creatinine estimation do not aid in its diagnosis [5]. Management modalities include creatine monohydrate supplementation and supportive rehabilitatory therapies, which retard the progression of disease but cannot reverse it [3].
X linked Mental Retardation (XLMR) comprise of a large heterogeneous group of disorders predominantly characterized by intellectual disability along with various co-morbidities occurring secondary to a mutation which is transmitted in X linked recessive manner [6].
This entity needs consideration in all cases of intellectual disability among males where major structural and metabolic etiologies and Fragile X syndrome have been excluded [6]. The clinical features in Case 3 were suggestive of a genetic etiology but in the absence of characteristic dysmorphism or clinical markers, could be diagnosed solely with the help of genetic testing. Genetic testing aids in classifying the etiology of intellectual disability and helps in antenatal counseling as in the index case [7]. Hereditary spastic paraplegia 47, as in Case 4, is a rare neurodegenerative disorder associated with neonatal hypotonia which evolves to spasticity of lower limbs along with severe intellectual disability [8]. This entity is one of the common mimics of cerebral palsy [9]. However, unlike cerebral palsy, it is a progressive disorder and can be genetically transmitted. Hence, thorough history and clinical examination followed by appropriate genetic testing, as in our index case, is mandated to provide appropriate prognosis and to prevent recurrence of similar illness in the family.
There is an expanding array of genetic tests, which aid in the antenatal diagnosis of developmental delay and intellectual disability [10]. Clinicians have a challenging role in advising genetic tests for intellectual disability due to the large number of genetic tests [11]. Appropriate history-taking and thorough clinical examination helps in choosing the most relevant test [11]. Antenatal counseling helps in the avoidance of recurrence of similar illnesses in family as in our case series.
Conclusion
Intellectual disability is a common entity, which can occur secondary to multiple etiologies. We report four cases of rare, genetically transmissible neurological disorders, which presented as intellectual disability. Appropriate genetic testing helped in offering antenatal counseling in all of these cases thereby highlighting the role of clinical examination and genetic testing in such scenarios.
Minor Thesis Statement
• Developmental delay and Intellectual disability are common clinical conditions that may occur secondary to multiple etiologies.
• Targetted investigations including genetic tests help in establishing the diagnosis of various inherited variants of Developmental Delay and Intellectual Disability as depicted through the reported cases.
• There is a definite role of antenatal genetic counselling in these disorders.
Source of Support
Nil.
Presentation at a Meeting
Nil.
Conflicting Interest
Nil.
Funds Received
Nil.
Acknowledgement
Nil.
Role of Contributors
RB : Case management, literature review,draft manuscript preparation, approval of final manuscript.
RDP: Case management,Interpretation of genetic reports,literature review,approval of final manuscript.
JNG: Clinician-in-charge,literature review,draft manuscript preparation, approval of final manuscript.
Contribution Details (to be ticked marked as applicable)
|
Contributor 1 (RB) |
Contributor 2 (RDP) |
Contributor 3 (JNG) |
|
|
Concepts |
☑ |
☑ |
☑ |
|
Design |
|
|
☑ |
|
Definition of intellectual content |
|
|
☑ |
|
Literature search |
☑ |
☑ |
☑ |
|
Clinical studies |
☑ |
☑ |
☑ |
|
Experimental studies |
|
|
|
|
Data acquisition |
☑ |
☑ |
|
|
Data analysis |
|
☑ |
☑ |
|
Statistical analysis |
|
|
|
|
Manuscript preparation |
☑ |
☑ |
☑ |
|
Manuscript editing |
☑ |
☑ |
☑ |
|
Manuscript review |
|
☑ |
☑ |
|
Guarantor |
|
|
☑ |
References
- Kishore MT, Udipi GA, Seshadri SP (2019) Clinical practice guidelines for assessment and management of intellectual disability. Indian J Psychiatry. 61: 194-210.
- Miclea D, Peca L, Cuzmici Z, et al. (2015) Genetic testing in patients with global developmental delay / intellectual disabilities. A review. Clujul Med 88: 288-292.
- Stockler S, Schutz PW, Salomons GS (2007) Cerebral creatine deficiency syndromes: Clinical aspects, treatment and pathophysiology. Subcell Biochem 46: 149-166.
- https://www.ncbi.nlm.nih.gov/books/NBK3794.
- Clark J, Cecil K (2015) Diagnostic methods and recommendations for the cerebral creatine deficiency syndromes. Pediatr Res 77: 398-405.
- Shevell M, Ashwal S, Donley D, et al. (2003) Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 60: 367-380.
- Ropers HH, Hoeltzenbein M, Kalscheuer V, et al. (2003) Nonsyndromic X‐linked mental retardation: Where are the missing mutations? Trends Genet 19: 316-320.
- Jamra A, Philippe R, Raas-Rothschild O, et al. (2011) Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet 88: 788-795.
- Pearson TS, Pons R, Ghaoui R, et al. (2019) Genetic mimics of cerebral palsy. Mov Disord 34: 625-636.
- Van den Veyver IB (2016) Recent advances in prenatal genetic screening and testing. F1000Res 5: 2591.
- Srour M, Shevell M (2014) Genetics and the investigation of developmental delay/intellectual disability. Arch Dis Child 99: 386-389.
Corresponding Author
Jyotindra Narayan Goswami, MD, DNB, DM, MNAMS, Associate Professor, Department of Pediatrics, Army Hospital (Research & Referral), Delhi Cantt, New Delhi,110010, India, Tel: 9893104114
Copyright
© 2021 Bhatt R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.