Reverse Somatic Mutations as Key Player in Malignant Transformation of an Odontogenic Myxoma
Introduction
Odontogenic myxoma (OM) is an uncommon neoplasm that affects the mandible or the maxilla and rarely occurs in other bones. It was first recognized in 1947 [1] with an annual incidence of 0.07 per million [2], likely derived from ectomesenchyme of the tooth germ. It commonly occurs in the 20-40s years of life with a wide age distribution from 3 months to 80 years and slightly female to male predominance with a ratio of 1.36:1 [3]. It is defined as an intraosseous benign but locally aggressive tumor, characterized by stellate and spindle -shaped cells dispersed in an abundant myxoid extracellular matrix [4], often being recurrent and bone destructive. However, due to the lack of in-depth genetic studies, it is unclear whether the recurrence and destruction are caused by malignant transformation or the expansive growing nature of the tumor itself. Some studies show that the local invasion may be related to increased expression of antiapoptotic protein Bcl2 and/or secretion of matrix metalloproteinases [5-7]. However, it is not clear whether malignancy also plays a role in local invasion. To address this concern, we studied longitudinally a series of tissue samples taken over a period of seven months from an OM patient by in-depth pathologic and next-generation sequencing analyses.
In recent comprehensive genetic studies on six different types of cancers [8,9], we identified that there were two opposite types of single nucleotide variations (SNVs) in both tumors and surrounding non-tumor tissues: One was gain-of-heterozygosity (GOH) type of mutation, in which a homozygous site was converted to a heterozygous site; another one was loss-of-heterozygosity (LOH) type of mutation, in which a heterozygous site was converted to a homozygous one. The LOH mutation is likely due to impaired DNA-damage response in form of double strand break repair by gene conversion [8]. GOHs and LOHs of single nucleotide were frequently found in tumors and adjacent non-tumor tissues [9]. Noticeably, these two types of SNVs were in mutually reversed status. If a forward mutation was GOH in a non-tumor tissue, then, the reverse mutations would be LOH in the neighboring tumor tissue; on the contrary, if a forward mutation was LOH in a non-tumor tissue, the reverse mutation would likely be GOH in the neighboring tumor tissue. Remarkably, more than half of the forward GOHs and LOHs occurring in non-tumor tissues would be reversed by the opposite LOHs and GOHs in malignant tumors. With these findings as a reference, we decided to see whether similar forward and reverse mutations occurred in this OM case that may substantiate a malignant transformation.
Materials and Methods
Case description
An 82-year-old Chinese male nonsmoker who had left maxillary pain and swelling started in 2016. He had occasionally bloodstained purulent discharge from the left side of the nose. Examination showed a hard palate bulging over the upper alveolar ridge with loosened left upper molar tooth that was badly eroded The CT scan showed an expansile growth of soft tissue mass in the left maxillary sinus. Biopses were performed. The first one was done on July 18, 2016, which showed only granulation tissue. The 2nd and 3rd biopsies were performed on August 3, 2016 and August 19, 2016 respectively, both of which displayed pauci cellularity of small spindle cells with tapering nuclei in a background of loose myxoid stroma. There were no mitotic figures or nuclear atypia. The tumor showed positive Alcian blue staining and Mib1 proliferation index was less than 5%. The diagnosis of OM was rendered. In view of clinical unusual growth, the fourth biopsy was done in early January, 2017, which showed markedly increased cellularity and mitotic figures with plumped spindle or stellate-shaped cells forming fascicles. The nuclei were pleomorphic and hyperchromatic. There was occasional cytoplasmic vacuoles. Proliferation index reached approximately 60%, which was highly indicative of malignant transformation, therefore, the tumor was surgically removed on January 27, 2017. The diagnosis of odontogenic myxoma with sarcomatous transformation was rendered. Postsurgical adjuvant radiotherapy was offered, but refused by the patient. Eight months later, in September, 2017, local recurrence was noted and radical maxillectomy was recommended, still he refused the procedure and the patient demised in May 2018. The biopsy specimens except first one and the surgical tumor tissue are used for genetic studies. The patient's white-blood-cell DNAs are used as the germline control.
Histopathological staining
Hematoxylin-eosin (HE), and Alcian blue histological staining of tumour sections were performed routinely in the pathology laboratory. Peripheral blood was collected from the patient with written consent. Previously recut unstained biopsy slides and portions of surgically removed tumor tissue were used for sequencing analysis.
Immunohistochemistry (IHC)
Surgically removed specimen was formalin fixed paraffin embedded and sectioned into 2 um unstained slides. Standard Polymeric Methods are utilized including antigen retrieval for following antibodies against vimentin (Dako), neuron specific enolase, Bcl2, AE1/3, MNF116 (Dako), SMA( IHC World), GFAP, S100, desmin, melanin A, CD34, ALK1 and CD117. OptiView DAB IHC detection kit was used for target detection with all slides stained on the VENTANA automated slide strainers and visualized by light microscopy with appropriate controls. All antibodies were products of Roche Tissue Diagnostics except a few listed above in parenthesis.
AluScan sequencing
An Alu element is about 300 base pairs, short interspersed nuclear elements (SINEs), characterized by the action of the Arthrobacter luteus (Alu) restriction endonuclease. There are over one million copies dispersed throughout the human genome. This abundance well positioned them to facilitate genome-wide sequence amplification. We used inter-Alu PCR with an enhanced range of amplicons in conjunction with next-generation sequencing to generate an Alu-anchored scan, or 'AluScan', of DNA sequences between Alu transposons, where Alu consensus sequence-based 'H-type' PCR primers that elongate outward from the head of an Alu element are combined with 'T-type' primers that elongate from the poly-A containing tail to achieve huge amplicon range. AluScan sequencing platform was used to detect genetic alterations in the patient's tumor tissue and peripheral blood. DNA extraction and AluScan sequencing library preparation were performed as described previously [10]. Genomic regions flanked by Alu repetitive sequences were obtained by means of inter-Alu PCR as described [10]. Both Head-type and Tail-type Alu consensus-based primers were used to ensure capture of a vast number of inter-Alu amplicons. In brief, a 25-μl PCR reaction mix containing 2 μl Bioline 10x NH4 buffer (160 mM ammonium sulfate, 670 mM Tris-HCl, pH 8.8, 0.1% stabilizer; www.bioline.com), 3 mM MgCl2, 0.15 mM dNTP mix, 1 unit Taq polymerase, and 0.1 μg DNA sample were prepared, and 0.075 μM each of the these were pipetted for PCR amplification. Alu consensus sequence-based PCR primers are listed as below: AluY278T18 (5'-GAGCGAGACTCCGTCTCA-3'); AluY66H21 (5'-GTCTCGATCTCCTGACCTC-3'); R12A/267 (5'-AGCGAGACTCCG-3'); L12A/8 (5'-TGAGCCACCGCG-3'). PCR was started at 95 ℃ 5 min for DNA denaturation, followed by 30 cycles, each of 30 s at 95 ℃, 30 s at 50 ℃, and 5 min at 72 ℃, plus another final 7 min at 72 ℃. Amplicons were purified with ethanol precipitation, sequenced on the Illumina HiSeq 2000 platform (San Diego, California, US) at Beijing Genomics Institute (Shenzhen, China) and mapped to the hg19 reference human genome for downstream bioinformatic analysis.
Gene network analysis
Network analysis was performed using the GeneMANIA Cytoscape plugin [11] software. A set of genes harboring non-synonymous mutations in the patient's germline genome were used as a query, and the gene functional association network was predicted accordingly. These functionally associated genes were further analyzed by the Reactome pathway database [12] software to identify the significantly enriched pathways.
Results
Description of radiology imaging findings
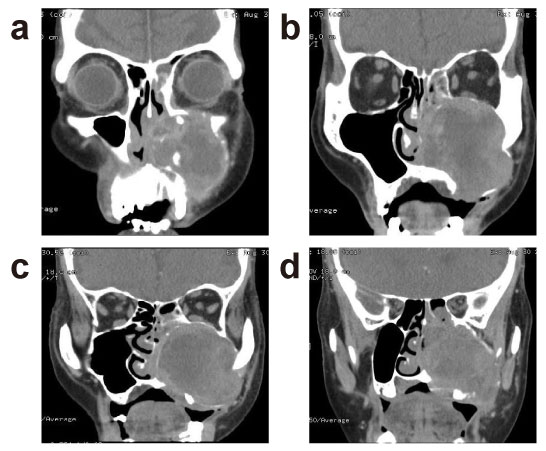
CT scan (Figure 1) demonstrated 5.3 × 5.3 cm mass in the left maxillary sinus with associated bony destruction, including inferior left orbital wall erosion, partially destruction of anterior maxillary wall, involvement of nasion, nasolacrimal duct, nasal cavity, choana, and ethmoidal sinus, pterygopalatine fossae, sphenopalatine foramen, alveolar process of maxilla, left side hard palate and the caudal part of the left inferior orbital fissure. The posterior wall of left maxilla appears intact. The pterygomaxillary fissure, rotundum, parapharyngeal fat spaces, and pterygoid muscles were spared.
Histopathological features of the tumor
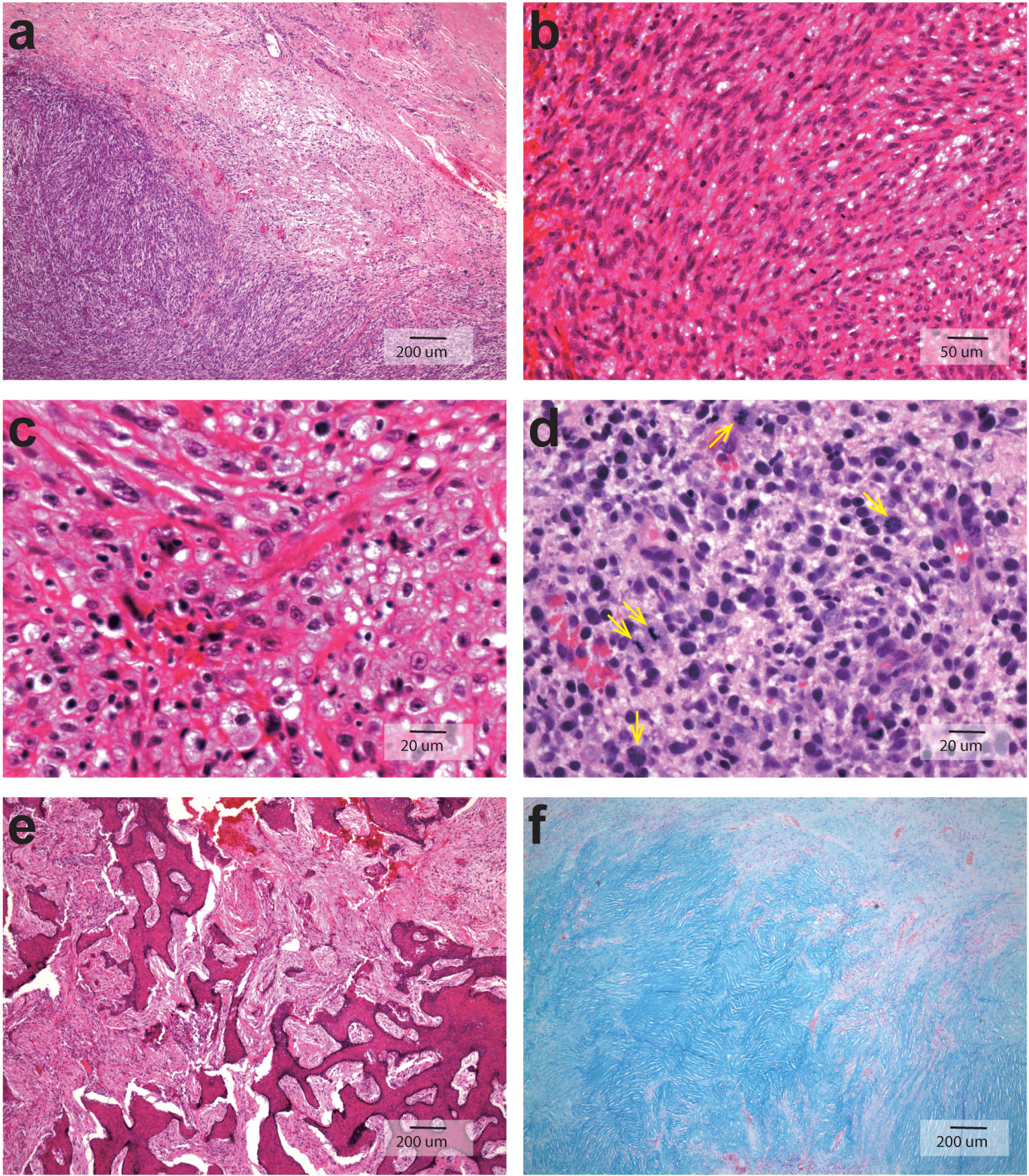
The tumor sections showed great variation in cellularity from low cellular to high cellular as displayed in Figure 2. There is sharp delineation between pauci cellular and hypercellular readily appreciated in Figure 2A, and Figure 2B showed predominant spindly tumor cell morphology. There were marked nuclear pleomorphism, hyperchromasia and frequent aberrant mitoses (pointed by arrows) as well as cytoplasmic vacuolization of the tumor cell as illustrated in Figure 2C and Figure 2D. Extensive fibrinoid necrosis and geographic hyalinization were also noted; the tumor cells infiltrated into the surrounding bony trabeculae, resulting in destruction and remodeling as demonstrated in Figure 2E; Figure 2F was Alcian blue staining on 2nd biopsy showing extracellular matrix stained positive for its abundant acid and neutral mucopolysaccharides, and this section showed pauci cellularity in myxoid stroma of benign odontogenic myxoma without atypia or mitoses.
IHC findings
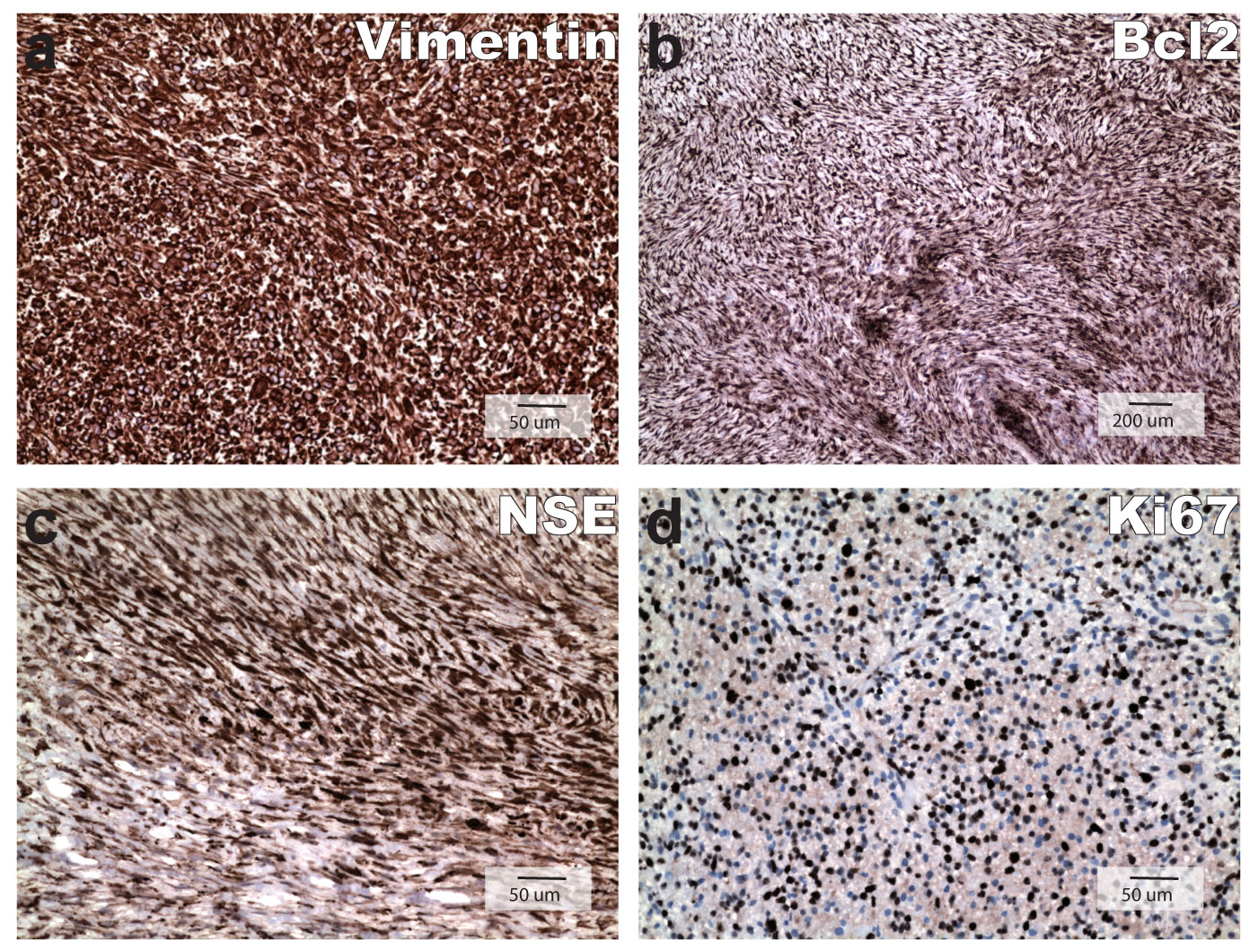
IHC stains of the tumor were strongly positive for vimentin, NSE, and Bcl2 and negative for AE1/3, MNF116, SMA, GFAP, S100, desmin, melanin A, CD34, ALK1 and CD117. A few mast cells are stained positive for CD117. Ki67 IHC stain showed a proliferation index approaching approximately 60%, which was similar to that of 4th biopsy specimen. Positive IHC staining for vimentin, Bcl2, NSE and Ki67 labeling were displayed as Figure 3 and all negative IHC stains were not shown.
Forward and reverse somatic mutations in benign and malignant tissues
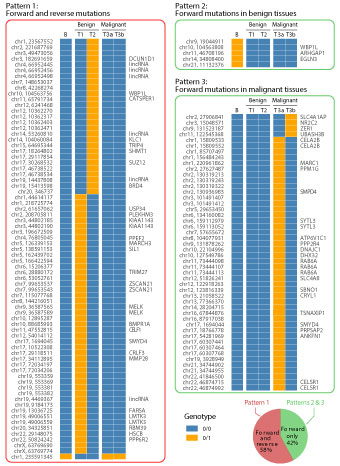
Four tissue specimens were analyzed for somatic SNVs. These included one sample each from the time-adjacent 2nd and 3rd biopsies designated as T1 and T2, as well as two pieces of tissue samples from different locations in the surgically resected tumor designated as T3a and T3b. The patient's white-blood-cell genomic DNA sample was designated as B and used as the germline control in next-generation sequencing analyses. Any base position in the tissue sample different from the blood control in genotype was considered as a somatic mutation in form of either GOH or LOH type of SNV. In comparison of T1, T2, T3a and T3b samples with B sample, forward mutations were identified if the genotypes in tumor tissue samples were different from the B sample, and reverse mutations were identified if the genotypes in T3a and T3b samples changed back to the genotypes in B sample. For instance, genotype 0/0 (blue rectangles in Figure 4) in blood changed to genotype 0/1 (orange rectangles in Figure 4) in a tumor sample was referred as forward GOH.
As illustrated in Figure 4, three different patterns of somatic mutational changes were observed when time serial tumor samples were compared with blood sample for genotypes at the same base positions. Pattern 1 was forward and reverse mutations. This pattern of mutation accounted for 58% of all somatic mutations. Most of these mutations were forward GOH (genotype 0/0 in blood changed to genotype 0/1 in benign tumor) followed by reverse LOH (genotype 0/1 in benign tissue changed to genotype 0/0 in malignant tumor); Pattern 2 was forward mutations observed in benign tumor, with all five being forward LOH mutations (genotype 0/1 in blood changed to genotype 0/0 in benign portion of the tumor), and the genotype remained the same in malignant tumor; pattern 3 was forward mutations observed in malignant tumor, which occurred only in tissue from benign to malignant transformation. The 2nd and 3rd patterns were similar in that both of them were forward mutations without reversals, and they jointly accounted for 42% of all somatic mutations. Notably, 62% somatic mutations were occurred in benign tumor. Also, T3a and T3b samples obtained from separate parts of the surgically removed tumor tissue were different in 38% of mutation sites (Pattern 3).
Germline mutations enriched in LRRFIP1 pathway
In order to determine the possible genetic predispositions of OM, germline mutations were identified from the patient's white-blood-cell genomic DNA sample. In the patient's white-blood-cell genome, a total of 47 non-synonymous SNVs in 41 genes have been detected, among which there were three nonsense mutations occurred in three genes, LRRFIP1, CA11 and TUBA8. And the remainders were missense mutations. The mutation data were summarized in Table 1. Non-synonymous germline mutations detected in the patient's peripheral blood.
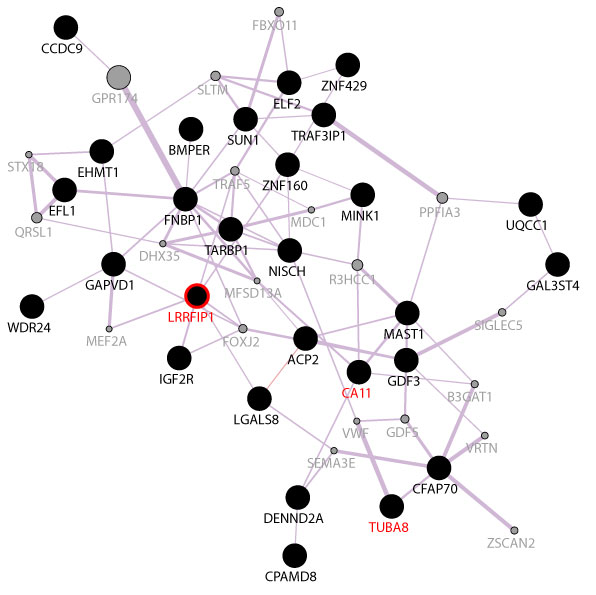
These 41 mutated genes were further analyzed using the GeneMANIA software to see if they were functionally correlated. All the genes that displayed non-synonymous mutations in the germline genome were imported to GeneMANIA for network analysis to determine whether these genes were reported to be functionally related in literatures. For example, the LRRFIP1 and ZNF160 were reported to show co-expression pattern by Smirnov, et al. [13]. In this way, we had identified that 28 out of the 41 genes were found previously to be co-expressed. The co-expression network of these genes was illustrated in Figure 5. The 28 co-expressed genes were analyzed with the Reactome pathway database [12], and the results showed that only LRRFIP1 activating type I interferon production pathway was enriched (Stable Identifier: R-HSA-3134973). LRRFIP1 is a cytosolic nucleic acid sensor gene, reported to be upregulated in breast cancer [14] and Burkitt lymphoma [15]. It also promotes colorectal cancer metastasis and liver invasion through integrin-dependent RhoA activation [16].
Discussion
The precise nature of OM is not entirely clear, especially its molecular pathogenesis. Local invasion can be seen in both benign and malignant OM cases, but distant metastasis has not been detected even in malignant one. The tumor growth is relatively slow, especially in benign ones. There are no specific IHC markers for OM, however, based on literature and our IHC results, OM is essentially all positive for vimentin and negative for keratin, desmin, GFAP, and S100 [17,18]. As for NSE, our result showed strong positivity in contrary to most negative reports, which were based on staining of benign OM. NSE has been shown to be positive in a wide range of mesenchymal and epithelial malignancies due to its heterodimers cross reaction. The sarcomatous transformation of OM in this patient may attribute its NSE IHC characteristics that not seen in benign OM. Bcl2, an anti-apoptotic protein has low expression in benign counterparts; its high expression in our case indicate that Bcl2 by preventing deliberate cell death has facilitated the tumor development. Very strikingly, proliferation index has increased from < 5% in benign component, which in line with literature reports [19-21], to close to 60% in sarcomatous component.
Apparently, OM with sarcomatous transformation is a rare phenomenon, few in literature reported. The first molecular-level malignant OM diagnosis was made based on hypertetraploidy [22]. A recently published NGS study of OM [23] failed to identify pathogenic mutations. Due to the scarcity of genetic data, we were not clear if a malignant OM start as a benign process and then evolved into malignancy or if it was a malignant tumor to begin with. By sectioning the entire surgically removed tumor in our patient, we noticed irregularly intermingled benign looking components with malignant ones indicating that the original OM was benign in nature and the tumor cells, through dedifferentiation transformed into a sarcoma. Our time series genomic sequencing analysis has confirmed this clinical observation.
In this patient, we detected a total of 47 non-synonymous germline mutations and 126 somatic mutations. The 47 non-synonymous germline mutations were enriched in the LRRFIP1 activating type I interferon production pathway. Previous studies found that type I interferon response was involved in innate immune sensing of tumor, and the damage to this pathway led to unfavorable clinical outcome in multiple types of cancers [24]. Since germline mutations also existed in somatic cells, the disturbance of this pathway may have played a role in malignant transformation of the tumor in this patient. Although the exact driver gene mutation cannot be definitively determined, we found that the genotypic changes in this patient's benign to malignant tumor transformation manifested similar characteristics of forward-and-reverse mutations pattern as we observed in our previous studies [9]. Our previous and current studies revealed that forward mutation occurred in the benign component is a prerequisite for a beginning stage of tumor development and reverse mutation is a prerequisite for the malignant development of the tumor [9]. It is likely that these forward mutations would increase the probability of generating essential mutations needed by the development of tumor cells and enabled them to further advance toward full-fledged malignancy. On the other hand, they could also bring out excessive mutation load that would be a burden for the tumor's growth. Thereby, to reduce mutation load, reverse mutations occurred in the genome of malignant tumors to render growth and survival advantages for the malignant cells.
The necessity of mutation load reduction in benign to malignant tumor transformation was also consistent with our previous findings by comparing N-, P- and T-stage cells [9]. The N-stage cells that were loaded with excessive forward mutations might be relatively deficient in growth and replication vigor until they accomplish their early mutation reversal in the P-stage and further mutation reversal in the T-stage. This series of reciprocal mutations empowered the functionality of the tumor to fully achieve malignant characteristics. Because the N-stage tissue contains numerous forward mutations, these tissue regions could be at premalignant or early malignant stage despite their apparently normal cytomorphology. These N-stage forward mutations were observed to undergo pronounced reversal in P- and T-stages, suggesting that the forward and reverse mutations during successive tumor development stages could be a common phenomenon, where tumor cells adopted evolution strategy to genetic changes encountered during their stage transitions.
This OM patient is the oldest among all OM patients reported. In general, OM cases are rarely seen in people older than 50s. The mandible appears to be more frequently affected than the maxilla. One reported malignant OM case which resulted in death occurred in maxilla in a patient over 50s [22]. Although there are no enough cases to substantiate it, old age and maxillary location should caution for potential malignant transition. To avoid sampling bias, different areas of tumor should be thoroughly sampled.
In conclusion, the occurrences of a wave of forward mutations followed by their reversals were observed in this patient in the process from benign to malignant transformation. Because tumors are driven by mutations, delineating the nature of mutations in the evolution process into malignant cells furnishes the basis for understanding carcinogenesis. In the present study, the analysis of time series samples in both benign and malignant tumor components from the same patient showed a bulk of forward mutations occurring in benign tissue were reversed in its malignancy. The dominance of reverse somatic mutations in the benign to malignant transformation is a cellular mechanism for gaining advantages of cell survival through regulation of the mutation load. The identification of reverse somatic mutations provides a basis for facilitating diagnosis of malignancy. Because this is a single case analysis, more genetic studies are needed to definitively elucidate genomic alterations driving the malignant transformation of OM and its enigma. The similar sequencing analysis of benign looking adjacent tissue should be performed in order to detect genomic changes when benign to malignant tumor transition is suspected.
Conflict of Interest Statements
The authors have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
References
- Thoma KH, Goldman HM (1947) Central myxoma of the jaw. Am J Oral SurgOrthod 33: 532-540.
- Simon EN, Merkx MA, Vuhahula E, et al. (2004) Odontogenic myxoma: a clinicopathological study of 33 cases. Int J Oral Maxillofac Surg 33: 333-337.
- Chrcanovic BR, Gomez RS (2019) Odontogenic myxoma: An updated analysis of 1,692 cases reported in the literature. Oral Dis 25: 676-683.
- (2017) Odontogenic myxoma/myxofibroma WHO classification of Head and Neck Tumors, 229.
- Miyagi SP, Hiraki KR, Martins MD, et al. (2008) Expression of matrix metalloproteinases 2 and 9 in odontogenic myxoma in vivo and in vitro. J Oral Sci 50: 187-192.
- Bast BT, Pogrel MA, Regezi JA (2003) The expression of apoptotic proteins and matrix metalloproteinases in ondotogenicmyxoma. J Oral Maxillofac Surg 61: 1463-1466.
- Mauro A, Lipari L, Tortorici S, et al. (2013) Expression of MMP-2 and MMP-9 in odontogenic myxoma in a child: report of a clinical case. Odontology 101: 233-238.
- Kumar Y, Yang J, Hu T, et al. (2015) Massive interstitial copy-neutral loss-of-heterozygosity as evidence for cancer being a disease of the DNA-damage response. BMC Med Genomics 8: 42.
- Hu T, Kumar Y, Shazia I, et al. (2018) Forward-reverse mutations in stages of cancer development. Hum Genomics 12: 40.
- Mei L, Ding X, Tsang SY, et al. (2011) AluScan: a method for genome-wide scanning of sequence and structure variations in the human genome BMC Genomics 12: 564
- Montojo J, Zuberi K, Rodriguez H, et al. (2014) GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Res 3: 153.
- Joshi-Tope G, Gillespie M, Vastrik I, et al. (2005) Reactome: a knowledgebase of biological pathways. Nucleic Acids Res 33: D428-D432.
- Smirnov DA, Morley M, Shin E, et al. (2009) Genetic analysis of radiation-induced changes in human gene expression. Nature 459: 587-591.
- Rikiyama T, Curtis J, Oikawa M, et al. (2003) GCF2: expression and molecular analysis of repression. Biochimica Biophysica Acta 1629: 15-25.
- Sjöblom T, Jones S, Wood LD, et al. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314: 268-274.
- Ariake K, Ohtsuka H, Motoi F, et al. (2012) GCF2/LRRFIP1 promotes colorectal cancer metastasis and liver invasion through integrin-dependent RhoA activation. Cancer Letters 325: 99-107.
- Lorenzo Lo Muzio, PierFrancesco Nocini, Gianfranco Favia, et al. (1996) Odontogenic myxoma of the jaws A clinical, radiologic, immunohistochemical, and ultrastructural study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 82: 426-433
- Tie-Jun Li, Li-Sha Sun, Hai-Yan Luo (2006) Odontogenic Myxoma: A Clinicopathologic Study of 25 Cases. Arch Pathol Lab Med 130: 1799-1806.
- Morihiro Higo, Atsushi Kasamatsu, Katsunori Ogawara, et al. (2015) A case of a rapidly expanding odontogenic myxoma of the mandibleOral Science International 12: 22-26.
- G Iezzi, A Piattelli, C Rubini, et al. (2007) Mib-1, Bcl-2 and p53 in odontogenic myxoma. ActaOtorhinolaryngol Ital 27: 237-242.
- Martínez-Mata G, Mosqueda-Taylor A, Carlos-Bregni R, et al. (2008) Odontogenic myxoma: clinico-pathological, immunohistochemical and ultrastructural findings of a multicentric series. Oral Oncol 44: 601-607.
- Pahl S, Henn W, Binger T, et al. (2000) Malignant odontogenic myxoma of the maxilla: case with cytogenetic confirmation. J Laryngol Otol 114: 533-535.
- JN dos Santos, S Neto, E Santos, et al. (2017) Next-generation sequencing of oncogenes and tumor suppressor genes in odontogenic myxomas. Journal of Oral pathology and Medicine 46: 1036-1039.
- Fuertes MB, Woo SR, Burnett B, et al. (2013) Type I interferon response and innate immune sensing of cancer. Trends Immunol 34: 67-73.
- Yates B, Braschi B, Gray KA, et al. (2017) Genenames.org: the HGNC and VGNC resources in 2017. Nucleic Acids Res 45: 619-625.
- Adzhubei IA, Schmidt S, Peshkin L, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248-249
Corresponding Author
Hong Xue, Division of Life Science, Applied Genomics Centre and Centre for Statistical Science, Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong; School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing, China.
Copyright
© 2019 Hu T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.