Malignant Peripheral Nerve Sheath Tumor with Loss of SMARCB1 and Diffuse S1000 Expression Arising in a Trigeminal Nerve Schwannoma
Abstract
Malignant peripheral nerve sheath tumors (MPNSTs) are rare sarcomas of the nerve sheath that infrequently involve the head and neck region. The few reports of MPNSTs arising from benign schwannoma precursors typically exhibit epithelioid morphology. We describe an exceptional case of a conventional MPNST arising in a benign trigeminal nerve schwannoma of a 21-year-old man without neurofibromatosis or familial schwannomatosis. Histologically, the tumor consisted of spindle-shaped cells in a fascicular and focal whorling growth pattern, with extensive geographic necrosis, cytologic atypia, rare atypical mitotic figures and focal histologic features of cellular schwannoma. Immunohistochemically, the tumor cells showed strong and diffuse S100 positivity and loss of SMARCB1. Subsequent genomic sequencing revealed a single nucleotide deletion (c.1148delC) in the SMARCB1/INI1 gene Diffuse S100 positivity and/or loss of SMARCB1 expression is extremely rare in conventional MPNST. We discuss diagnostic considerations for MPNSTs and review the literature concerning treatment of these rare neoplasms.
Keywords
Malignant peripheral nerve sheath tumor, SMARCB1/INI1, Malignant transformation, Schwannoma
Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are rare sarcomas that arise from cells associated with peripheral nerve sheaths. They constitute approximately 5-10% of all soft tissue sarcomas, with most MPNSTs occurring in the torso and extremities and only 8-16% in the head and neck region [1]. MPNSTs can occur spontaneously, though up to 50% are associated with neurofibromatosis type 1 and develop either de novo or from a preexisting neurofibroma [2]. Malignant transformation of schwannomas is exceedingly rare, with only 20 reported cases [3], and usually shows epithelioid morphology [4].
SMARCB1 is a tumor suppressor gene whose mutation has been described in the malignant rhabdoid tumor of infancy, and has more recently been linked to the pathogenesis of additional tumors, including a subset of epithelioid MPNSTs [5]. However, to our knowledge there has not been a report examining SMARCB1 inactivation in a conventional spindle cell MPNST.
Here we report a conventional MPNST with loss of SMARCB1and diffuse S100 expression arising in a trigeminal nerve schwannoma of a young man without neurofibromatosis or familial schwannomatosis.
Clinical History
A 21-year-old previously healthy male presented with a one-month history of worsening right-sided nasal obstruction and two weeks of diplopia and right-sided facial numbness. He reported occasional bloody mucus but denied overt epistaxis. Nasal endoscopy showed a right-sided nasal mass with superficial vascularity in the middle meatus. MRI and CT showed a 58 mm mass that appeared to be centered along the right foramen rotundum and involving the right sinonasal cavity, nasopharynx, right orbit and multiple right skull base foramina, with intracranial extension into the inferior lateral right cavernous sinus and Meckel’s cave. Positron emission tomographic scanning showed no evidence of metastasis. Initial biopsy was obtained in clinic under diagnostic sinonasal endoscopy and pathological diagnosis was spindle cell neoplasm, favor cellular schwannoma. However, based on the aggressive clinical presentation and extensive local involvement, the initial biopsy was believed to not be representative of the lesion. Therefore, a second biopsy was performed in the operating room and the pathologic diagnosis was malignant peripheral nerve sheath tumor. The patient underwent two cycles of doxorubicin and ifosfamide. Repeat MRI at two months following initial presentation showed a slight increase in tumor size. The patient continued to be symptomatic, and was thus taken to the operating room for surgical debulking. Compared to its appearance at the time of second biopsy, the tumor was noted to have increased vasculature and extension. Currently, the patient is undergoing two cycles of ifosfamide and etoposide. We will then restage him and consider further chemotherapy or consolidative radiation therapy.
Pathology
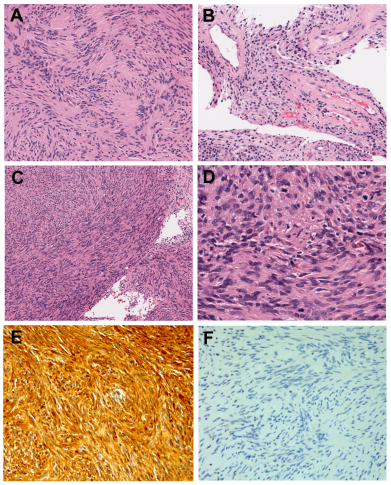
Grossly, the initial biopsy specimen consisted of a 1.5 × 1.0 × 0.3 cm aggregate of pink-red irregular soft tissue fragments. Microscopically, haematoxylin and eosin (H&E) stain showed sheets of cellular bland spindle cell proliferation in a fascicular growth pattern intermingled with clots. Tumor cells were narrow, elongate, and wavy with tapered ends, ill-defined cytoplasm and dense chromatin interspersed with collagen fibers. Occasional nuclear palisading around a fibrillary process (Verocay body) was seen (Figure 1A). Gaping tortuous lumina with thickened hyalinized walls were focally present (Figure 1B). There was no necrosis and no mitotic figures were identified. Upon immunohistochemical staining, tumor cells were diffusely and strongly immunoreactive for S100, while negative for AE1/3, EMA and CD34. Based on the morphologic appearance as described above and the immunophenotypic profile, initial pathologic diagnosis was spindle cell neoplasm, favor cellular schwannoma.
Larger fragmented materials from the second biopsy were examined. On H&E stain, similarly to initial biopsy, the tumor was cellular and had a fascicular and focal whorling growth pattern (Figure 1C); however, there was extensive geographic necrosis. The tumor cells were spindle, ovoid, or serpentine-shaped and were larger and plumper compared to the initial biopsy, with cytologic atypia (Figure 1D). There were rare mitotic figures (approximately 3/10 high power fields). The tumor cells showed diffuse and strong S100 immunoreactivity (Figure 1E), and a Ki-67 proliferative index of approximately 10%. The final diagnosis was malignant peripheral nerve sheath tumor, grade 2 (FNCLCC grade system, Fédération Nationale des Centres de Luttele Cancer).
The tumor cells in the initial biopsy and the re-biopsy material showed virtual loss of SMARCB1 expression by immunohistochemical study (Figure 1F). Genomic DNA was extracted from the second biopsy according to manufacturer’s instructions (Qiagen, Inc). Targeted analysis for mutations in the regions specified in this testing panel was achieved by enrichment of those genomic loci using the Illumina Truseq Amplicon Assay. Sequencing of enriched libraries was performed on the Illumina MiSeq platform using multiplexed, paired end reads. Analysis and interpretation utilized a customized bioinformatics process. The sequencing panel consisted of 47 genes (ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL). The tumor sample demonstrated a deletion of a single cytosine nucleotide in the SMARCB1 gene (c.1148delC) with an allele frequency of 39.64%, inducing a frame shift after Pro383 (p.P383Rfs*100).
Discussion
MPNSTs are rare neoplasms that have a total incidence of approximately 0.001% and tend to present during mid-adulthood [1,6,7]. While benign schwannomas have a propensity for cranial nerves, MPNSTs of the cranial nerves are exceptionally rare [6]. According to a literature review by Schmidt, et al., only 36 patients with trigeminal nerve MPNSTs have been previously reported.
Overall, the diagnosis of MPNST is difficult and can necessitate a combination of clinical, radiological and pathological findings. Clinical presentation of MPNSTs can resemble that of benign schwannomas, but features of large size, rapid enlargement, pain and neurological deficits are more predictive of malignancy [8,9]. Most common clinical manifestations of trigeminal MPNSTs include altered facial sensation and facial pain, although involvement of other cranial nerves can lead to additional symptoms [6]. The presenting symptoms of facial numbness, diplopia and nasal obstruction in our patient resulted from the tumor’s anatomic location and regional extension.
Radiologically, it can also be difficult to distinguish MPNSTs from their benign counterparts such as neurofibromas and schwannomas. Common features include a spindle shape and longitudinal orientation to the nerve distribution. However, MRI findings more suggestive of malignancy include large size (> 5 cm), invasion of fat planes, heterogeneity, ill-defined margins, peripheral enhancement, perilesional edema and adjacent bone destruction [9,10].
Specific morphological, immunohistochemical and molecular criteria for MPNST are lacking. Upon gross examination, MPNSTs are usually white, solid and fleshy lesions that frequently exhibit myxoid change, necrosis and hemorrhage [11]. Microscopically, MPNSTs can display a variety of cell morphologies, including spindle, epithelioid, pleomorphic or small round cell. They exhibit diverse growth patterns, including a branching hemangiopericytoma-like vascular pattern, alternating hypercellular and hypocellular areas, and a whirling or rarely palisading growth pattern. The tumor cells may have fibroblastic features or heterologous elements such as skeletal muscle, bone, cartilage and blood vessels, or rarely glandular differentiation with or without mucin production. It can be associated with geographic necrosis, which may be extensive [4,11]. The differential diagnosis of MPNST is broad, as pathological appearance can resemble that of other sarcomas such as benign schwannoma, neurofibroma, fibrosarcoma, melanoma and other spindle cell sarcomas [4,12]. It is particularly challenging to diagnose in a small biopsy, as demonstrated in the present case.
Immunohistochemical S100 staining is variable and usually only focal, correlating with the degree of Schwannian differentiation, and GFAP staining is only positive in 20-30% of cases [11]. Diffuse S100 positivity in MPNST is generally seen only in pediatric cases associated with neurofibromatosis [13]. Epithelioid MPNST, a rare variant, accounts for approximately 5% of cases and is characterized by clusters of large, rounded cells with prominent nucleoli. In contrast to conventional - MPNST, epithelioid MPNST typically show strong and diffuse S100 protein expression [4,11]. Lack of SMARCB1 expression has also been associated with epithelioid MPNSTs, with a recent series of epithelioid sarcomas demonstrating that 50%~67% of epithelioid MPNSTs showed loss of SMARCB1 [5,14], which has not been reported in conventional MPNST. The genetic alteration in SMARCB1 in epithelioid PMNST is unknown. The SMARCB1/INI1 gene is localized to chromosome 22q11.2 and encodes the SNF5 protein, one of three core subunits of the SWI/SNF family of chromatin remodeling complexes. SNF5 plays an important role in epigenetic regulation and cell cycle control [13,15]. In addition to epithelioid PMNST, SMARCB1 loss has been reported in malignant rhabdoid tumors, and more recently in a variety of additional malignancies including epithelioid sarcoma, renal medullary carcinoma, medullary carcinoma, extraskeletal myxoid chondrosarcoma, familial schwannomatosis, and rare subset of sinonasal carcinoma [5,15-17].
The majority of MPNSTs develop de novo in normal peripheral nerves or from neurofibroma precursors, primarily the plexiform type in the setting of neurofibromatosis 1 [11]. Only rare examples of MPNST arise from schwannoma, ganglioneuroma and pheochromocytomas [7,11], though relatively higher associations with schwannomas have been reported in cranial nerve MPNSTs [12]. While neurofibromas usually give rise to conventional spindle cell MPNSTs, schwannomas tend to give rise to sarcomas with epithelioid features [18].
The present case demonstrated conventional MPNST spindle cell morphology with focal features of cellular schwannoma with diffuse and strong S100 staining in the original in-clinic biopsy. The initial diagnosis favor cellular schwannoma but given the aggressive clinical course and radiologic appearance a second biopsy of the 58 mm mass was acquired, which showed spindle cell proliferation with fascicular and focal whorling growth. The main differentiation is cellular schwannoma, however, clinical presentations and histologic features including extensive geographic necrosis, cytologic atypia, and rare atypical mitosis are consistent with PMNST. The discrepancy between the two biopsies suggests the tumor’s heterogeneity and development from a benign schwannoma. Interestingly, the tumor showed strong and diffuse S100 positivity, SMARCB1/INI1 single nucleotide deletion and loss of SMARCB1/INI1 expression. Both strong and diffuse S100 staining and lack of SMARCB1 expression are common in epithelioid PMNST, but extremely unusual in conventional - MPNST. We found only one reported case of conventional spindle cell-type malignant peripheral nerve sheath tumor arising in a sporadic schwannoma, in which SMARCB1 protein expression was preserved [18]. Loss of SMARCB1 expression in epithelioid MPNST specifically arising in schwannoma has been reported in a single case [3]. However, to our knowledge, SMARCB1 mutation in a conventional spindle cell MPNST has not been described. As such, our case represents the first report of a SMARCB1 deficient spindle cell MPNST, as well as the second report of a conventional MPNST arising in a sporadic benign schwannoma [18].
Surgical resection is the mainstay of treatment for MPNSTs, as complete surgical excision with wide negative margins has been shown to the most important prognostic factor [6,19,20]. However, this is particularly difficult to achieve in MPNSTs of the head and neck given the region’s anatomic complexity and close proximity to critical structures. Radiotherapy has shown to be essential for local disease control for most sarcomas and likewise can serve as adjunctive treatment for MPNST. Although MPNSTs have historically been thought to be radioresistant, more recent reports suggest that radiation therapy offers decreased incidence of local recurrence and may have a role as primary therapy for unresectable lesions [6,20-22]. The benefit of radiotherapy on long-term survival is still debated [21,23].
The role of chemotherapy in MPNST remains poorly defined, with only occasional reports of favorable responses to chemotherapy [20-22]. Doxorubicin and ifosfamide in combination with surgery and radiotherapy has been shown to have a survival benefit in a group of patients with large and high-grade MPNSTs [24], but other studies have not found significant improvement in survival [20,22,25,26]. More recently, a phase II clinical trial (NCT00304083, SARC006) evaluated the role of chemotherapy with doxorubicin, ifosfamide and etoposide in patients with unresectable or metastatic MPNST. Results indicated disease stabilization and partial decrease in tumor size in most patients [27].
Rates of local recurrence and distant metastasis tend to be high, and documented 5-year survival rates for MPNSTs of the head and neck have ranged from 15% to 35% [20]. Factors that predict survival include tumor size, surgical margin status, tumor location and histologic grade [1,20,22,27].
Extension into the cavernous sinus, internal carotid artery and the orbit precluded complete surgical resection of our patient’s tumor. Therefore, the multidisciplinary team at our institution elected to pursue chemotherapy following the SARC006 clinical trial protocol [27], which the patient is currently undergoing.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- Patel TD, Shaigany K, Fang CH, et al. (2016) Comparative analysis of head and neck and non-head and neck malignant peripheral nerve sheath tumors. Otolaryngol Head Neck Surg 154: 113-120.
- Evans DG, Baser ME, McGaughran J, et al. (2002) Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 39: 311-314.
- Carter JM, O'Hara C, Dundas G, et al. (2012) Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with "neuroblastoma-like" schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol 36: 154-160.
- Rodriguez FJ, Folpe AL, Giannini C, et al. (2012) Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 123: 295-319.
- Hornick JL, Dal Cin P, Fletcher CD (2009) Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33: 542-550.
- Schmidt RF, Yick F, Boghani Z, et al. (2013) Malignant peripheral nerve sheath tumors of the trigeminal nerve: A systematic review of 36 cases. Neurosurg Focus 34: E5.
- Ren X, Wang J, Hu M, et al. (2013) Clinical, radiological, and pathological features of 26 intracranial and intraspinal malignant peripheral nerve sheath tumors. J Neurosurg 119: 695-708.
- Furniss D, Swan MC, Morritt DG, et al. (2008) A 10-year review of benign and malignant peripheral nerve sheath tumors in a single center: Clinical and radiographic features can help to differentiate benign from malignant lesions. Plast Reconstr Surg 121: 529-533.
- Pilavaki M, Chourmouzi D, Kiziridou A, et al. (2004) Imaging of peripheral nerve sheath tumors with pathologic correlation: Pictorial review. Eur J Radiol 52: 229-239.
- Yu YH, Wu JT, Ye J, et al. (2016) Radiological findings of malignant peripheral nerve sheath tumor: Reports of six cases and review of literature. World J Surg Oncol 14: 142.
- Thway K, Fisher C (2014) Malignant peripheral nerve sheath tumor: Pathology and genetics. Ann Diagn Pathol 18: 109-116.
- Scheithauer BW, Erdogan S, Rodriguez FJ, et al. (2009) Malignant peripheral nerve sheath tumors of cranial nerves and intracranial contents: A clinicopathologic study of 17 cases. Am J Surg Pathol 33: 325-338.
- Zhou H, Coffin CM, Perkins SL, et al. (2003) Malignant peripheral nerve sheath tumor: A comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am J Surg Pathol 27: 1337-1345.
- Jo VY, Fletcher CD (2015) Epithelioid malignant peripheral nerve sheath tumor: Clinicopathologic analysis of 63 cases. Am J Surg Pathol 39: 673-682.
- Kalimuthu SN, Chetty R (2016) Gene of the month: SMARCB1. J Clin Pathol 69: 484-489.
- Agaimy A, Koch M, Lell M, et al. (2014) SMARCB1 (INI1)-deficient sinonasal basaloid carcinoma: A novel member of the expanding family of SMARCB1-deficient neoplasms. Am J Surg Pathol 38: 1274-1281.
- Bishop JA, Antonescu CR, Westra WH (2014) SMARCB1 (INI-1)-deficient carcinomas of the sinonasal tract. Am J Surg Pathol 38: 1282-1289.
- Endo M, Yamamoto H, Harimaya K, et al. (2013) Conventional spindle cell–type malignant peripheral nerve sheath tumor arising in a sporadic schwannoma. Hum Pathol 44: 2845-2848.
- Zou C, Smith KD, Liu J, et al. (2009) Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 249: 1014-1022.
- Anghileri M, Miceli R, Fiore M, et al. (2006) Malignant peripheral nerve sheath tumors: Prognostic factors and survival in a series of patients treated at a single institution. Cancer 107: 1065-1074.
- Minovi A, Basten O, Hunter B, et al. (2007) Malignant peripheral nerve sheath tumors of the head and neck: Management of 10 cases and literature review. Head Neck 29: 439-445.
- Mullins BT, Hackman T (2014) Malignant peripheral nerve sheath tumors of the head and neck: A case series and literature review. Case Rep Otolaryngol 2014: 368920.
- Kahn J, Gillespie A, Tsokos M, et al. (2014) Radiation therapy in management of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Front Oncol 4: 324.
- Moretti VM, Crawford EA, Staddon AP, et al. (2011) Early outcomes for malignant peripheral nerve sheath tumor treated with chemotherapy. Am J Clin Oncol 34: 417-421.
- Kolberg M, Holand M, Agesen TH, et al. (2013) Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol 15: 135-147.
- Pfeiffer J, Arapakis I, Boedeker CC, et al. (2008) Malignant peripheral nerve sheath tumour of the paranasal sinuses and the anterior skull base. J Craniomaxillofac Surg 36: 293-299.
- Widemann B, Reinke D, Helman LJ (2013) SARC006: Phase II trial of chemotherapy in sporadic and neurofibromatosis type 1 (NF1)-associated high-grade malignant peripheral nerve sheath tumors (MPNSTs). J Clin Oncol 31: a10522.
Corresponding Author
Melissa Zheng, BA, School of Medicine, University of California-San Francisco, 513 Parnassus Ave, San Francisco, CA 94143, USA.
Copyright
© 2019 Zheng M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.