Sox2 Functions in Neural Cancer Stem Cells: The Importance of the Context
Abstract
The Sox2 transcription factor is expressed in different neural tumors. In particular, it is active within the "cancer stem cell" (CSC) subpopulation of tumor cells, able to reinitiate tumorigenesis after conventional chemotherapy (to which it is usually resistant). This led to hypothesize that Sox2 (and its downstream regulated genes) may qualify as promising targets for therapeutic strategies directed against CSC. However, the potential relevance of Sox2 in this regard depends on whether it is functionally important to maintain CSC. Here, we comparatively examine the effects of Sox2 genetic ablation within mouse models of different neural tumor types. Sox2 ablation in mouse glioma (and in human glioblastoma-derived CSC) demonstrated a critical function for Sox2 in the maintenance of CSC. Surprisingly, however, Sox2 ablation in two different mouse models of melanoma (a neural crest-related tumor), and in a mouse model of medulloblastoma of the Sonic Hedgehog subgroup, showed that, in these contexts, Sox2 is dispensable for tumorigenesis. This heterogeneous situation has a parallel in the normal development of the nervous system, where generalized Sox2 ablation in neural stem/progenitor cells selectively affects the development of some neural regions, but not other ones. Molecular mechanisms underlying these specificities may involve the regulation, by Sox2, of different sets of target genes in different tumors, but also a redundant regulation of the same targets by different Sox transcription factors, differentially coexpressed with Sox2 in different tumors. Collectively, these findings point to the need to experimentally address the requirement for Sox2, and its downstream targets, within different tumor types, as a prerequisite to fully exploit its potential as a target for novel therapeutic approaches.
Keywords
Sox2, Cancer stem cells, Cancer, Glioblastoma, Oligodendroglioma, Medulloblastoma, Melanoma, Mouse genetic models
Abbreviations
CSC: Cancer Stem Cells; ESFT: Ewing Sarcoma Family Tumors; MB: Medulloblastoma; NSC: Neural Stem Cells; SCC: Squamous Cell Carcinoma; SCLC: Small Cell Lung Carcinoma
Introduction
Stem cells are "cells that have the ability to perpetuate themselves through self-renewal and to generate mature cells of a particular tissue through differentiation" [1]. "Cancer stem cells" (CSC) were identified in human brain tumors [3-3], as a minority subpopulation able to reinitiate tumor development following conventional chemotherapy (to which they are usually resistant), or, experimentally, following transplantation in a host (mouse) brain. Much evidence has accumulated supporting the importance of CSC for tumor relapse and propagation [2,4,5], and CSC are now considered a central target for therapeutic approaches aimed at eradicating tumor development. CSC self-renew, and also produce "pseudo-differentiated" cells, constituting the tumor bulk [6-8]. While most of the tumor bulk cells are typically proliferating (though they are called "differentiated" from the CSC perspective), CSC can be slowly proliferating and even "quiescent" [6,8], making the use of drugs hitting proliferating cells uneffective in killing them.
Sox2 is a member of the Sry-related HMG-box (SOX) family of transcription factors; the Sox2 gene is located on chromosome 3, in both humans and mice, and in both species it is constituted by a single coding exon [9]. Sox2 was first studied in the context of normal embryonic development, where knock-out experiments in mouse demonstrated its essential role to maintain the pluripotent stem cells of the early embryo [10], as well as its function in several tissue-specific stem cells, including neural stem cells (NSC); NSC cultured in vitro from the mutant neonatal mouse brain fail to self-renew in long-term culture, and, in vivo, postnatal hippocampal NSC are impaired [11-14].
The discovery of CSC in tumors focused attention on Sox2 also from the point of view of this pathological stem cell type. Indeed, Sox2 is expressed in different tumors of neural origin, such as gliomas (the most common primary brain tumors, whose most malignant and lethal subtype is glioblastoma multiforme), medulloblastomas (the most common brain tumor in childhood), and melanoma (a tumor arising from neural crest type cells); In all of these tumors, CSC have been identified, and found to express Sox2 [2,8,15].
In this paper, we focus on the comparative review of recent experiments, that made use of conditional mutation of Sox2 in different mouse models of neural tumors (and of genetic Sox2 ablation in human CSC-enriched cultures of the same tumor types), to investigate the functional relevance of Sox2 in tumorigenesis, and, in particular, in the maintenance of CSC. These experiments demonstrated an absolute requirement for Sox2 of CSC able to reinitiate tumorigenesis of gliomas (glioblastoma and oligodendroglioma) in mouse; unexpectedly, however, they also showed that Sox2 is dispensable for tumorigenesis in a model of Sonic Hedgehog (SHH)-subgroup medulloblastoma, and in two different models of melanoma. We discuss a parallel of this situation with the identification of region-specific Sox2 functions in the context of the development of the normal neuroepithelium, and possible molecular mechanisms underlying the context-specificity of Sox2 functions. Finally, we discuss the need and approaches to identify functional downstream effectors of Sox2 in Sox2-dependent CSC, that could complement Sox2 as targets of therapeutic strategies directed against CSC.
Sox2-dependent neural cancer stem cells in gliomas
Gliomas are the most common cerebral neoplasias (86%) [16-18]. Glioblastoma multiforme (GBM), the most aggressive and deadly among gliomas (average patients� survival is about 15 months), was one of the first tumor types in which CSC were originally described (reviewed in [2]. The development of serum-free in vitro cultures of tumor-derived cells has allowed to expand CSC (in equilibrium with more differentiated progeny) from GBM tissue of many different patients; these cells retain the ability to re-form a tumor with the same characteristics of the tumor of origin following transplantation into a host mouse brain (xenograft), and thus represent important in vitro models of CSC. GBM, and CSC-enriched cultures derived from it, consistently express Sox2 [19].
Gangemi, et al. [20] first addressed the consequences of lowering Sox2 levels in some patient-derived CSC-enriched cell cultures, by expressing anti-Sox2 shRNAs causing strong reduction of Sox2 mRNA levels. This resulted in reduced cell proliferation and reduced clonogenicity in vitro, and to loss of tumorigenicity in vivo, in mouse xenografts [20]. A role for Sox2 in maintaining other patient-specific GBM CSC was further supported in additional important work by other laboratories [15].
In mouse cells, complete Sox2 ablation was obtained in vitro by conditional knockout in a genetically defined model of glioma: a high-grade oligodendroglioma induced by overexpression of Platelet-derived growth factor B (PDGF-B) [21]. Oligodendroglioma is the second most common tumor in adults; patients with high-grade oligodendroglioma have a median survival of 3-4 years [16,22]. Alterations of PDGF-B signalling are common molecular lesions in human gliomas, including oligodendrogliomas: PDGF and PDGF receptor have both been found overexpressed in glial tumor-derived cells and glioma surgical samples, and amplification of the gene encoding the PDGF receptor-A occurs in high-grade oligodendrogliomas [23-25]. In addition, PDGF can initiate the "reprogramming" of normal, committed O2A oligodendrocyte progenitors to neural stem-like cells, in a process that requires Sox2 [26]. PDGF-B was used to induce tumor development in embryonic brain; cells cultured from such tumors would re-initiate tumor development following transplantation, thus behaving as CSC. Sox2 ablation was achieved by conditional knock-out, in which Cre recombinases delete the endogenous Sox2 gene, that has been previously flanked by loxP sites, the Cre substrate ("Sox2 flox" allele) [13]. Sox2 Cre-mediated ablation in vitro in such glioma cells completely prevented tumor reinitiation following in vivo transplantation; over the time window in which non-deleted cells developed deadly tumors, mice transplanted with Sox2-deleted cells remained tumor free. The only two tumors developing from Cre-treated cells were SOX2-positive, demonstrating they were derived from non-deleted cells. in vitro, Sox2 ablation caused proliferation reduction, apoptosis activation, and aberrant differentiation into cells expressing oligodendrocyte and astrocyte markers [21]. Counterintuitively, Sox2, or peptide fragments of it, though being a nuclear protein, can be found exposed on the cell surface of tumor cells, in association with the Major Histocompatibility Complex (MHC), presumably as a result of tumor cell lysis and processing of its proteins; based on this, immunotherapy against SOX2 protein was attempted, which resulted in a doubling of survival time of mice transplanted with oligodendroglioma cells [21].
Sox2 cooperating genes
Recent experiments investigated transcription factors cooperating with Sox2 in cell "reprogramming" to tumor-propagating cells (cancer stem cells). In recent work [7], SOX2 was able to reprogram "differentiated" GBM cells from human tumors (DCGs) to stem-like tumor-propagating cells (TPC), when transduced into the DCGs together with transcription factors POU3F2 (BRN2), SALL2, and OLIG2. In GBM-derived CSC-enriched cultures grown in serum-free conditions, genomic mapping (by ChIPseq) of H3K27Ac, a histone modification carried by active enhancers and promoters, had initially revealed regulatory elements specific of TPC (versus DCGs), and these were enriched in the DNA sequence recognized by SOX proteins. RNA seq showed that expression of SOX2 (and of SOX1, SOX5, SOX8, SOX21) was higher in TPC than in DCGs ("differentiated" by serum or BMP4 addition). Similarly, transcription factors SALL2, POU3F, and OLIG2 were selected based on two considerations: enrichment, in TPC versus DCGs, of their expression, and the presence of H3K27Ac (an epigenetic mark of transcriptionally active state) on their binding sites on gene promoters and enhancers. The combination of SOX2 (but not SOX1), SALL2, POU3F and OLIG2 (or Rest Corepressor 2, RCOR2, a transcriptional corepressor) could reprogram DCGs to TPCs, that carried a genome-wide pattern of H3K27Ac sites superimposable to that of the TPCs that had been directly grown from the tumor. Further, the 4 factors co-bind to a large number of distal regulatory elements specifically active in TPCs. Interestingly, a minority of cells coexpressed the 4 transcription factors in the tumor, as demonstrated by immunofluorescence. Looking for therapeutic implications, two mediators acting downstream to the 4 transcription factors were also identified: RCOR2 (that can replace OLIG2 in the reprogramming cocktail) and histone demethylase LSD1, whose repression caused cell death specifically in TPCs. Interestingly, it had previously been found that LSD1-specific inhibitors impaired the growth of Sox2-expressing, but not that of Sox2-negative, lung squamous cell carcinomas (SCC), and Sox2 expression was associated with sensitivity to LSD1 inhibition in lung, breast, ovarian, and other carcinoma cells [27]. This indicates that LSD1 is a mediator of tumorigenic effects downstream to Sox2, but not other tumorigenic factors.
In a related study, ChIPseq in GBM cells showed that DNAase hypersensitive sites in patient-derived GBM cells, mapped by ATAC-seq (a technique that allows to map DNA regions that are more accessible in chromatin, usually in correlation with the binding of transcription factors), are enriched in DNA recognition sequences for SOX2 and FOXG1, a transcription factor active in embryonic brain development; ectopic expression of SOX2 and FOXG1 in postmitotic astrocytes reactivated proliferation and stem cell properties. The coexpression of FOXG1 with SOX2 in GBM led to hypothesize that they could, together, contribute to cell "reprogramming" to stemness in gliomagenesis [28].
Sox2 relevance for non-neural tumors. Common downstream Sox2 effectors?
Importantly, conditional knockout and RNA interference experiments showed that SOX2 controls CSS functions also in some non-neural tumours, such as in skin, lung, and aesophagus squamous cell carcinomas (SCC), osteosarcomas, Ewing sarcoma, and small cell lung carcinomas (SCLC) [29-33]. This suggests that some downstream effectors of Sox2 function may be conserved between different tumors. In this regard, in osteosarcoma-initiating cells, Sox2 directly represses the genes encoding two activators of the "hippo" signalling pathway, Nf2 (Neurofibromin 2, also called Merlin, encoding a protein involved in connecting the cytoskeleton with proteins of the cell membrane) and WWC1 (also called Kibra, encoding a cytoplasmic phosphoprotein), which in turn negatively regulate the transcriptional coactivator YAP, important for promoting tumor growth; interestingly, Sox2 depletion led to upregulation of Nf2 and WWC1, downregulation of YAP, and to reduced cell clonogenicity, in both osteosarcoma and GBM cells, indicating that these effectors may be shared between these tumor types [30]. In osteosarcomas, as well as in laryngeal cancer cells, Sox2 was also proposed to control migration and invasion via the Wnt/beta-catenin signalling pathway [34,35], though enforced Sox2 expression in lung adenocarcinoma was reported to promote cell migration and invasion, but to inhibit Wnt/beta-catenin signalling activity [36]. Thus, Sox2 might regulate the Wnt/beta-catenin pathway in different tumor types, though not necessarily with the same functional outcome. In hematopoietic tumors, Sox2 expression was detected in cultured cells isolated from ALK-positive anaplastic large cell lymphoma, and Sox2 downregulation impaired their clonogenicity and tumorigenic ability; oxidative stress increased Sox2 expression and cancer stem cell properties in a subpopulation of cells, and Sox2 was reported to bind DNA more efficiently [37]. It is possible that specific partners had become available in oxygen-stressed cells, or that increased levels of Sox2 were important for binding; alternatively, posttranslational modifications of Sox2 were proposed to be involved in this "activation" following oxidative stress [38].
Interestingly, some papers reported Sox2 expression and some functional effects of Sox2 downregulation also in human mammary cancer cell lines [39,40]. However, in a widely studied mouse model of breast cancer, produced by expression of a transgene encoding a mutated ErbB2/Neu oncogene driven to mammary tissue by the MMTV promoter [41,42], and activated by a mammary-specific Cre recombinase [43], Sox2 ablation had no effect on tumorigenesis (A.B.S. and S.K.N., unpublished observations). It is possible that breast tumors are heterogeneous regarding functional requirements for Sox2 in tumorigenesis. Indeed, Sox2 expression was observed in some breast tumors (mostly belonging to early stages of tumor progression), but not in others, and some, but not all breast tumor-derived stem cell-enriched cultured cell lines are reported to express Sox2 [39]. A future characterization of the gene regulatory networks acting in breast cancer CSC might allow to better categorize them with respect to Sox2 function.
It is interesting to note that Sox2 is important also in the normal cell counterparts of CSC within several of the non-neural tissues (see above) known to develop Sox2-dependent tumors, as previously seen with NSC [13]: indeed, normal osteoblasts, dermal papilla cells, and cells of the developing foregut (giving rise to lung, esophagus and trachaea) require Sox2 function [4-46]. This indicates that Sox2 function, already present in tissue-specific stem/progenitor cells, is retained by CSC of (at least some) tumors of the same tissue type. A different situation was documented in Ewing Sarcoma Family Tumors (ESFT), mesenchimal tumors thought to arise from primary mesenchymal stem cells. Here, SOX2 expression is strongly activated de novo by the oncogenic transcription factor that characterizes a high proportion of ESFT, encoded by the EWS-FLI-1 fusion gene. EWS-FLI-1 activates SOX2 (together with OCT4 and NANOG), and SOX2 is a key factor in the emergence of a ESFT CSC population; its downregulation in ESFT cells antagonizes cell proliferation and tumorigenesis [32].
Collectively, these findings show that SOX2 is required by CSC in various gliomas, in mouse and human (as well as by CSC of several non-neural cell types). This raises the possibility that SOX2 may qualify as a target for CSC-directed therapy strategies; note that, though Sox2 is highly expressed in gliomas and CSC, its expression is very limited in normal brain tissue surrounding the tumor. They further indicate that the identification of new downstream mediators of Sox2 function in gliomas may be of relevance for therapy approaches, not only in glioma, but also in other Sox2-dependent tumor types, where some relevant Sox2-controlled gene regulatory networks may be conserved (see also the recent reviews on the subject by [15,47].
Medulloblastoma Development in a Sox2-ablated Mouse Model
Medulloblastoma (MB), the most common brain tumor in childhood, was shown early on to harbour CSC [2,3]. Quite some studies were devoted to the Sonic Hedgehog (SHH) subgroup of MB, representing about 30% of total MB, presenting aberrant SHH signalling because of loss of function of negative regulators (including PTCH1, SUFU), activating mutations of positive transducers (SMO), or amplification of transcriptional effectors, like GLI2 [48]. SHH subgroup MB were shown to originate from cerebellar granule neuron precursors, that proliferate during normal development under the physiological stimulus of SHH [49]. In these tumors, SHH pathway inhibitors entered clinical trials, but reports of resistance and relapse indicate the possibility that an insensitive CSC might be spared [8].
In mouse, a model of the SHH subgroup MB is the irradiated Ptch +/- mouse, where postnatal irradiation increases tumor frequency from 20% to 80%. Recent work in this model has shown that rare, quiescent cells, expressing Sox2, behave as tumor-propagating cells following transplantation, and in primary tumors in situ [8]. Sox2-expressing cells, and their progeny, were labelled in vivo through activation of a GFP transgene by an inducible Cre (CreERT2) driven by the Sox2 locus, and characterized through tumor development, by immunofluorescence. This revealed that rare Sox2-positive cells (less than 5% of total) produce rapidly proliferating progenitors (marked by doublecortin, DCX), that, together with their non-dividing progeny (positive for NeuN) constitute the tumor bulk. The fraction of Sox2-positive cells increased following anti-mitotic, or anti-SMO, therapy, which kills dividing cells; this suggests that Sox2-positive cells were spared by these therapies and could be responsible for the observed relapse. These findings indicate Sox2-positive cells as a promising target for anti-CSC therapy in SHH subgroup MB but leave open the question of whether Sox2 itself is functionally relevant for them.
Sox2 function in mouse MB tumorigenesis has been tested by conditional Sox2 knockout within another model of SHH subgroup MB, the lox-stop-lox-SmoM2-YFP mouse [50]. SmoM2 encodes a mutated version of the Smo gene, encoding the SHH co-receptor; SmoM2 was originally discovered as an activating mutation of Smo (ant thus of the SHH pathway) in basal cell carcinomas [51]. In the mouse model, the SmoM2 transgene activates SHH signalling following Cre recombinase-mediated excision of the stop sequence [49]. In hGFAPCre:SmoM2flox/+ mice, SmoM2 is activated throughout the neuroepithelium, and MB develops [49]; these MB expresses Sox2, with sparse cells showing particularly high expression levels [51]. In hGFAPCre:Sox2flox/flox:SmoM2flox/+ mice, tumors developed, where SOX2 was not detected. However, the development and morphology of the tumors were not significantly altered by Sox2 loss; further, the survival time was not significantly different between Sox2-positive and Sox2-negative genotypes.
It should be noted that, in this model, SHH signalling is activated uniformly within the many cells in which SmoM2 is activated by Cre, which does not mirror the pathological situation in which an oncogenic mutation is first present in just one cell. However, in this model, Sox2 ablation did not preclude MB tumorigenesis. It is possible that, in MB, Sox2 is upstream to genes regulating SHH signalling, as seen in normal NSC [13]; in the SmoM2 model, Sox2 function might be bypassed by autonomous, constitutive activation of the SHH pathway.
In this model, expression of Sox3, a transcription factor belonging to a subgroup of Sox genes coexpressed with Sox2 in the developing neuroepithelium [12], was detected in the tumor, and found upregulated in mutant cerebella following Sox2 ablation [51]. Sox transcription factors co-bind to many target genes [52], so these findings raise the possibility that Sox3 acts redundantly with Sox2 to maintain MB tumorigenesis. Vice versa, pro-differentiative Sox factors (Sox5, 6, 21) were found to be downregulated during malignant glioma progression, their genetic ablation increased the ability of cells to form glioma-like tumors, and expression of high levels of Sox5/6/21 in primary human GBM cells antagonized their tumorigenic capacity [53]. A similar, antagonistic function of Sox9 and Sox10 was also described in melanoma [54] (see below Figure 1). It is possible that the Sox genes coexpressed with Sox2 in tumors and CSC may represent a generally important factor conditioning Sox2 function in CSC. Their importance may be tested by genetic ablation experiments, in combination with Sox2.
Sox2-independent Neural Cancer Stem Cells in Melanoma
Melanoma is a skin tumor originating from malignant transformation of melanocytes, cells derived from the embryonic neural crest [55]. About 50% of human melanomas express Sox2 [56]; further, Sox2 has functional roles in normal melanocyte progenitors of the developing neural crest, as Sox2 gradual downregulation permits the differentiation of neural crest (and Schwann cells)-derived melanocyte progenitors into melanocytes [57,58]. Sox2 has thus been considered a candidate to play functional roles also in melanoma. A different Sox factor, Sox 10, had been previously shown by conditional knock-out to play an essential role in melanoma development within a mouse genetic model of NrasQ61K-driven melanomagenesis [59], prompting related experiments for Sox2.
The study of Sox2 function in human melanoma-derived cells by RNAi-mediated knockdown approaches had given controversial results. Whereas some Sox2 knockdown experiments indicated a contribution of Sox2 to the maintenance of patient-derived melanoma spheres self-renewal and xenograft tumor development [60], and Sox2 overexpression increased melanoma cell invasion [61], in other reports Sox2 knockdown failed to elicit effects [56]. In more recent experiments, Sox2 was fully ablated via CRISPR/Cas9-mediated mutagenesis in human patient-derived melanoma cells carrying a N-Ras Q61L oncogenic mutation and expressing high Sox2 levels [58]. Following xenotransplantation into immunocompromised mice, Sox2-deleted cells were equally capable of generating tumors than non-deleted cells [58], pointing to a dispensable role for Sox2.
In mouse, Sox2 function in in vivo melanomagenesis was recently studied in two different genetic models of melanoma, by conditional Sox2 knockout. The first model is the Tyr:CreERT2 :: NRasQ61K Ink4a-/- mouse, in which a transgene (Tyr:CreERT2), expressing the inducible CreERT2 in melanocytes, is coupled to a Cre-activatable transgene, carrying the oncogenic NRasQ61K mutation, together with a homozygous mutation of Ink4a [58]. This model recapitulates all phases of melanomagenesis from benign nevi formation from melanocytes, to malignant transformation, metastases, and tumor dissemination at distant sites; in this model, the requirement for Sox10 in melanomagenesis had previously been demonstrated [59]. In the second model, a BrafV600E mutation cooperates with Pten loss to induce melanoma [62]. Both tumors express Sox2, at least in a fraction of cells. In both cases, Sox2 ablation was obtained by combining these oncogenic mutations with a homozygous Sox2flox allele; this caused Sox2 deletion, by the same Cre recombinases that activated the oncogenic transgenes. In both cases, Sox2 ablation did not prevent tumor formation, and the kinetics and characteristics of tumor growth did not differ detectably in Sox2-negative and Sox2-positive (control) tumors, nor did the survival; further, to test for Sox2 requirement in different phases of tumorigenesis, deletion was induced before the appearance of primary melanomas, or when metastases formed, and again no difference was observed with controls, despite efficient Sox2 deletion [58,62]. This indicates that, at least in these models, Sox2 is dispensable for tumor development from melanocytes. It is possible that the different findings in some human cell lines and in the two in vivo mouse models (see above) reflect a previously unappreciated heterogeneity in melanomas regarding Sox2 requirement. Functional experiments will be important in discriminating, among melanomas, those requiring Sox2 function for tumorigenesis.
A Parallel: Different Requirements for Sox2 by Stem/Progenitor Cells of Different Regions of the Normal Developing Nervous System
It may be interesting to note that Sox2 function is highly context-dependent also in the development of the normal nervous system. Following Sox2 deletion throughout the developing neural tube at embryonic day 11.5 (E11.5) (via a nestin-Cre transgene), development of the hippocampus is severely perturbed, whereas neural development in general is comparatively spared [13]. Following even earlier Sox2 ablation throughout the developing telencephalon, at E9.5 (via a FoxG1Cre transgene), the ventral telencephalon (medial ganglionic eminences, the primordium of the basal ganglia) is essentially lost, as is the olfactory neuroepithelium, whereas the dorsal telencephalon (cortex primordium) is comparatively less affected [58,64]. Sox2 ablation in the developing midbrain/hindbrain led to impaired development of the cerebellar vermis, and of postnatal cerebellar Bergmann glia, but not (or much less) of other cerebellar regions and cell types [65]. These findings indicate that Sox2 normally functions in a stage-, region-, and cell type-specific way in neural cells, driving context-specific gene regulatory networks.
Conclusions and Perspective
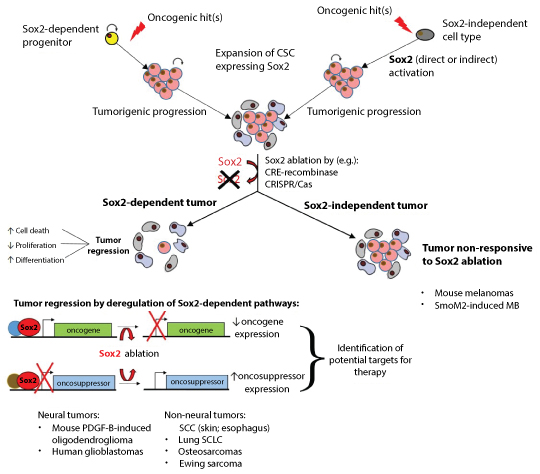
We have summarized evidence showing that Sox2 is essential for the maintenance of CSC and tumorigenesis in some neural tumor types, while being dispensable in others.
What molecular mechanisms could be involved in this context-specificity of functions in CSC? It is possible that different stem cell programs control the maintenance of, for example, glioma versus melanoma stem cells, and that only the first one requires Sox2. Alternatively, it is possible that, at least in some cases, Sox2 acts redundantly with other Sox factors, coexpressed with it in some tumor types (e.g. Sox3 in MB, see above). The combined ablation, in model systems, of the Sox genes potentially involved, by conditional knockout or CRISPR-Cas-mediated mutagenesis, should clarify these points.
On the other hand, we noted that a requirement for Sox2 is found not only in neural CSC within gliomas, but also in very different, non-neural tumors, such as skin and esophagus SCC, lung SCLC, and osteosarcomas. Perhaps, although they differ by histology and by cell of origin, these tumors share a "core", Sox2-controlled gene regulatory network, active in their CSC. Thus, it will be important to comparatively characterize the gene regulatory networks controlled by Sox2 in these CSCs.
These findings have implications for therapy approaches. On one hand, they suggest it might be advantageous to "classify" tumours according to the gene regulatory networks that function in the maintenance of their CSC, that in turn might involve shared efficacy of CSC-targeting drugs. On the other hand, they emphasize the need for functional experiments, to address the importance of specific gene products (here, Sox2, and its downstream targets), to distinguish driver from bystander roles, in order to appropriately target future CSC-directed therapy approaches.
Acknowledgements
Work in the Nicolis laboratory has been supported by Associazione Italiana per la Ricerca sul Cancro (AIRC-IG), the European Community (ERANET-NEURON ImprovVision), Telethon, Fondazione Cariplo. We wish to thank Sergio Ottolenghi for insightful discussion.
References
- Reya T, Morrison SJ, Clarke MF, et al. (2001) Stem cells, cancer, and cancer stem cells. Nature 414: 105-111.
- Nicolis SK (2007) Cancer stem cells and "stemness" genes in neuro-oncology. Neurobiol Dis 25: 217-229
- Singh SK, Hawkins C, Clarke ID, et al. (2004) Identification of human brain tumour initiating cells. Nature 432: 396-401.
- Beck B, Blanpain C (2013) Unravelling cancer stem cell potential. Nat Rev Cancer 13: 727-738.
- Pattabiraman DR, Weinberg RA (2014) Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov 13: 497-512.
- Chen J, Li Y, Yu TS, et al. (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488: 522-526.
- Suva ML, Rheinbay E, Gillespie SM, et al. (2014) Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157: 580-594.
- Vanner RJ, Remke M, Gallo M, et al. (2014) Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer cell 26: 33-47.
- Kondoh H, Robin LB (2016) Sox2, biology and role in development and disease. Elsevier, Associated Press.
- Avilion AA, Nicolis SK, Pevny LH, et al. (2003) Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17: 126-140.
- Arnold K, Sarkar A, Yram MA, et al. (2011) Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9: 317-329.
- Bertolini J, Mercurio S, Favaro R, et al. (2016) Sox2-dependent regulation of neural stem cells and CNS development. In: Kondoh H, Lovell-Badge R, Sox2, Biology and role in development and disease. Elsevier.
- Favaro R, Valotta M, Ferri AL, et al. (2009) Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat Neurosci 12: 1248-1256.
- Pevny LH, Nicolis SK (2010) Sox2 roles in neural stem cells. Int J Biochem Cell Biol 42: 421-424.
- Garros-Regulez L, Garcia I, Carrasco-Garcia E, et al. (2016) Targeting Sox2 as a therapeutic strategy in glioblastoma. Front Oncol 6: 222.
- Lau CS, Mahendraraj K, Chamberlain RS (2017) Oligodendrogliomas in pediatric and adult patients: an outcome-based study from the surveillance, epidemiology, and end result database. Cancer Manag Res 9: 159-166.
- Rasmussen BK, Hansen S, Laursen RJ, et al. (2017) Epidemiology of glioma: clinical characteristics, symptoms, and predictors of glioma patients grade I-IV in the danish neuro-oncology registry. J Neurooncol 135: 571-579.
- Reifenberger G, Wirsching HG, Knobbe-Thomsen CB, et al. (2017) Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol 14: 434-452.
- Lee J, Kotliarova S, Kotliarov Y, et al. (2006) Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer cell 9: 391-403.
- Gangemi RM, Griffero F, Marubbi D, et al. (2009) SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 27: 40-48.
- Favaro R, Appolloni I, Pellegatta S, et al. (2014) Sox2 is required to maintain cancer stem cells in a mouse model of high-grade oligodendroglioma. Cancer Res 74: 1833-1844.
- Reis-Filho JS, Faoro LN, Carrilho C, et al. (2000) Evaluation of cell proliferation, epidermal growth factor receptor, and bcl-2 immunoexpression as prognostic factors for patients with World Health Organization grade 2 oligodendroglioma. Cancer 88: 862-869.
- Appolloni I, Calzolari F, Tutucci E, et al. (2009) PDGF-B induces a homogeneous class of oligodendrogliomas from embryonic neural progenitors. Int J Cancer 124: 2251-2259.
- Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, et al. (2006) PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 51: 187-199.
- Shih AH, Holland EC (2006) Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer lett 232: 139-147.
- Kondo T, Raff M (2004) Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev 18: 2963-2972.
- Zhang X, Lu F, Wang J, et al. (2013) Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep 5: 445-457.
- Bulstrode H, Johnstone E, Marques-Torrejon MA, et al. (2017) Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev 31: 757-773.
- Bass AJ, Watanabe H, Mermel CH, et al. (2009) SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 41: 1238-1242.
- Basu-Roy U, Seo E, Ramanathapuram L, et al. (2012) Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene 31: 2270-2282.
- Boumahdi S, Driessens G, Lapouge G, et al. (2014) SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511: 246-250.
- Riggi N, Suva ML, De Vito C, et al. (2010) EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev 24: 916-932.
- Rudin CM, Durinck S, Stawiski EW, et al. (2012) Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 44: 1111-1116.
- Tang L, Wang D, Gu D (2018) Knockdown of Sox2 inhibits OS cells invasion and migration via modulating wnt/beta-catenin signaling pathway. Patholo oncol res 24: 907-913.
- Yang N, Hui L, Wang Y, et al. (2014) Overexpression of SOX2 promotes migration, invasion, and epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in laryngeal cancer Hep-2 cells. Tumour Biol 35: 7965-7973.
- He J, Shi J, Zhang K, et al. (2017) Sox2 inhibits Wnt-beta-catenin signaling and metastatic potency of cisplatin-resistant lung adenocarcinoma cells. Mol med rep 15: 1693-1701.
- Wu C, Gupta N, Huang YH, et al. (2018) Oxidative stress enhances tumorigenicity and stem-like features via the activation of the Wnt/beta-catenin/MYC/Sox2 axis in ALK-positive anaplastic large-cell lymphoma. BMC cancer 18: 361.
- Gupta N, Gopal K, Wu C, et al. (2018) Phosphorylation of Sox2 at threonine 116 is a potential marker to identify a subset of breast cancer cells with high tumorigenecity and stem-like features. Cancers 10.
- Leis O, Eguiara A, Lopez-Arribillaga E, et al. (2012) Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 31: 1354-1365.
- Wu F, Zhang J, Wang P, et al. (2012) Identification of two novel phenotypically distinct breast cancer cell subsets based on Sox2 transcription activity. Cellular signal 24: 1989-1998.
- Guy CT, Webster MA, Schaller M, et al. (1992) Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A 89: 10578-10582.
- Muller WJ, Sinn E, Pattengale PK, et al. (1988) Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54: 105-115.
- Wagner KU, Wall RJ, St-Onge L, et al. (1997) Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res 25: 4323-4330.
- Basu-Roy U, Ambrosetti D, Favaro R, et al. (2010) The transcription factor Sox2 is required for osteoblast self-renewal. Cell death and differentiation 17: 1345-1353.
- Clavel C, Grisanti L, Zemla R, et al. (2012) Sox2 in the dermal papilla niche controls hair growth by fine-tuning BMP signaling in differentiating hair shaft progenitors. Dev cell 23: 981-994.
- Domyan ET, Ferretti E, Throckmorton K, et al. (2011) Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development 138: 971-981.
- Wuebben EL, Rizzino A (2017) The dark side of SOX2: Cancer - a comprehensive overview. Oncotarget 8: 44917-44943.
- Northcott PA, Jones DT, Kool M, et al. (2012) Medulloblastomics: The end of the beginning. Nat Rev Cancer 12: 818-834.
- Schuller U, Heine VM, Mao J, et al. (2008) Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer cell 14: 123-134.
- Ahlfeld J, Favaro R, Pagella P, et al. (2013) Sox2 requirement in Sonic hedgehog-associated medulloblastoma. Cancer Res 73: 3796-3807.
- Xie J, Murone M, Luoh SM, et al. (1998) Activating smoothened mutations in sporadic basal-cell carcinoma. Nature 391: 90-92.
- Bergsland M, Ramsköld D, Zaouter C, et al. (2011) Sequentially acting Sox transcription factors in neural lineage development. Genes Dev 25: 2453-2464.
- Kurtsdotter I, Topcic D, Karlen A, et al. (2017) SOX5/6/21 Prevent oncogene-driven transformation of brain stem cells. Cancer Res 77: 4985-4997.
- Shakhova O, Cheng P, Mishra PJ, et al. (2015) Antagonistic cross-regulation between Sox9 and Sox10 controls an anti-tumorigenic program in melanoma. PLoS Genet 11: e1004877.
- Shakhova O (2014) Neural crest stem cells in melanoma development. Curr Opin Oncol 26: 215-221.
- Laga AC, Zhan Q, Weishaupt C, et al. (2011) SOX2 and nestin expression in human melanoma: An immunohistochemical and experimental study. Exp dermatol 20: 339-345.
- Adameyko I, Lallemend F, Furlan A, et al. (2012) Sox2 and Mitf cross-regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 139: 397-410.
- Schaefer SM, Segalada C, Cheng PF, et al. (2017) Sox2 is dispensable for primary melanoma and metastasis formation. Oncogene 36: 4516-4524.
- Shakhova O, Zingg D, Schaefer SM, et al. (2012) Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat cell biol 14: 882-890.
- Santini R, Pietrobono S, Pandolfi S, et al. (2014) SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 33: 4697-4708.
- Girouard SD, Laga AC, Mihm MC, et al. (2012) SOX2 contributes to melanoma cell invasion. Laboratory investigation 92: 362-370.
- Cesarini V, Guida E, Todaro F, et al. (2017) Sox2 is not required for melanomagenesis, melanoma growth and melanoma metastasis in vivo. Oncogene 36: 4508-4515.
- Ferri A, Favaro R, Beccari L, et al. (2013) Sox2 is required for embryonic development of the ventral telencephalon through the activation of the ventral determinants Nkx2.1 and Shh. Development 140: 1250-1261.
- Panaliappan TK, Wittmann W, Jidigam VK, et al. (2018) Sox2 is required for olfactory pit formation and olfactory neurogenesis through BMP restriction and Hes5 upregulation. Development 145.
- Cerrato V, Mercurio S, Leto K, et al. (2018) Sox2 conditional mutation in mouse causes ataxic symptoms, cerebellar vermis hypoplasia, and postnatal defects of Bergmann glia. Glia.
Corresponding Author
Silvia K Nicolis, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milano, Italy.
Copyright
© 2018 Barone C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.