NSAIDs, Dehydration and Acute Kidney Failure in the Young Adults: An Unrecognized Entity?
Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used drug classes worldwide. As a side effect of the therapy can occur an acute interstitial nephritis (AIN) or an acute tubular necrosis (ATN). In acute kidney injury, a major challenge is to distinguish between underlying etiologies. For instance, there are no disease-specific therapies for acute tubular necrosis, whereas acute interstitial nephritis requires immunosuppressive therapy.
The differentiation of the two diseases is very important, but unfortunately, they present clinically and laboratory similarly. Good biomarker for differential diagnosis is lacking. Often renal biopsy is required for diagnosis. Renal biopsy is often associated with side effects and cannot be performed in all patients. Therefore, it is very important to know the small differences of the two diseases. Enclosed we present three typical cases in which we diagnosed ATN based on the clinic, history, and laboratory findings.
Keywords
Acute interstitial nephritis, Acute kidney injury, Acute tubular injury, Nonsteroidal anti-inflammatory drugs, Nephrotoxicity
Abbreviation
AIN: Acute Interstitial Nephritis; AKI: Acute Kidney Injury; ATI: Acute Tubular Injury; BOLD MRI: Blood Oxygenation Level-Dependent Magnetic Resonance Imaging; CRP: C-Reactive Protein; COX: Cyclooxygenase; COX-1: Cyclooxygenase 1; COX-2: Cyclooxygenase 2; LE: Leukocytes; NSAIDS: Nonsteroidal Anti-Inflammatory Drugs; OTC: Over-the-Counter; PG: Prostaglandin; pO2: Partial Pressure of Oxygen; RBC: Red Blood Cell; RTEC: Renal Tubular Epithelial Cell; TNF-α: Tumor Necrosis Factor Alpha; WBC: White Blood Cell -/+ - Not present or only slightly increased; +/++ - Slightly to moderately elevated
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used drug classes worldwide and the estimated usage of NSAIDs are over 30 million people per day [1-3]. They are prescribed in medical practice as analgesics, antipyretics and anti-inflammatories and are considered safe and well tolerated [1,2].
NSAIDs selectively or preferentially inhibit COX-2 or non-selectively inhibit both COX-1 and COX-2 enzymes [4]. The different classes of NSAIDs have different potencies, which, in addition to their clinical efficacy, also affects their side effects and explains their ability to cause hypersensitivity reactions [4].
The dose and duration of use of NSAIDs therapy affects the pharmacological action [1,2,5]. They can cause both acute and chronic renal failure and in addition to acute tubular necrosis and interstitial nephritis, NSAIDs can cause glomerular lesions, decreased sodium and water excretion, hyperkalemia, renal tubular acidosis and hypertension [1,6]. NSAID therapy can also cause gastrointestinal, hepatic, cardiovascular side effects, intracerebral haemorrhage, respiratory tract complications and alter the platelet function [2,6,7].
In pediatrics, several case reports or small series of children have been described who have suffered an AKI after taking an NSAID. Frequently, these patients had gastroenteritis and were volume-depleted [8]. In healthy adults, the incidence of adverse renal effects is very low (< 1%); however, the elderly are at higher risk. Elderly patients may have age-related decrease in glomerular filtration rate, diabetes, congestive heart failure, hypovolemia due to use of loop diuretics, liver cirrhosis, pathological conditions leading to worsening of renal function or alteration of pharmacokinetics of NSAID [1].
Cases
Case 1
A 19-year-old woman was referred to the emergency department by her primary care physician for bilateral flank pain, elevated creatinine, and elevated inflammatory marker. She was taking amoxicillin/clavulanic acid, ibuprofen and metamizol for three days following tooth extraction. Three days after initiation of the therapy recurrent nausea, vomiting and bilateral flank pain occurred. The primary care physician initiated ciprofloxacin the same day for suspicion of urinary tract infection in a patient with a medical history of recurrent urinary tract infection. The urine status and urine culture remained without any findings during the course of the treatment. The extended medical history and medication history were unremarkable.
On admission, the patient was afebrile, cardio-pulmonary stable and in good general condition. The patient did not suffer from myalgia, arthralgia, fatigue or headache and the remaining systemic history was unremarkable. The clinical examination was unremarkable apart from a kidney tenderness on percussion on both sides. The laboratory analysis revealed an acute kidney injury (AKI II) and elevated CRP without leukocytosis or eosinophilia. The urine analysis remained unremarkable, and the sodium excretion fraction was 2.3%. Ultrasound examination off the kidneys reviled bilaterally enlarged kidneys with symmetrically elevated resistance indices (RI) as a sign of intrinsic renal disease. The examination excluded obstruction, renal congestion, and evidence of pyelonephritis.
The diagnosis of the acute kidney injury with acute tubular necrosis triggered by prior NSAID use and intermittent dehydration was retained. A postrenal cause was excluded at the emergency department. An acute glomerulonephritis was unlikely in the absence of proteinuria and bland urine sediment. Tubulointerstitial nephritis was excluded as unlikely because there was no evidence of allergic reaction or systemic involvement, and an unremarkable urine status with absence of leukocyturia, of eosinophilia and of albuminuria. Enlarged kidneys at ultrasound and flank pain were suggestive of acute tubular necrosis. The antibiotic therapy was stopped and intravenous combined with peroral hydration were given. Renal retention parameters rapidly declined. One week after hospitalization renal function was fully recovered.
Case 2
A 25-year-old man presented to the emergency department after several hot summer days because of right-sided flank pain with radiation to the right lower abdomen. The patient complained of nausea, vomiting, and decreased drinking volume. Due to the pain, the patient took unclear amount of ibuprofen. In medical history, the patient reported pollen allergy, nicotine consumption, a Bosniak 1 cyst of the left kidney, and an episode of febrile urinary tract infection one year prior to this hospitalization. The patient did not take medication regularly.
At presentation, the patient was afebrile, cardio-pulmonary stable and in good general condition. The clinical examination revealed reduced bowel sounds, soft abdomen, positive McBurney and psoas signs on the right side without peritonitis signs and a renal tenderness on percussion on the right side. The laboratory chemistry showed a slight increase in inflammatory parameters, acute kidney injury (AKI II) and absence of eosinophilia. The first urine examination showed an unclear proteinuria of 1.8 g/L and ketonuria as a sign of mild starvation. There was no evidence of urinary tract infection, leukozyturia or erythrocyturia. The young patient was hospitalized. Appendicitis, kidney stones and a postrenal cause were excluded by abdominal ultrasound and CT abdomen. Under fluid substitution and painkiller therapy, the patient's pain improved rapidly, and renal values decreased.
There was no evidence of glomerulonephritis in the absence of erythrocyturia and with rapidly regressing proteinuria. No systemic involvement or eosinophilia were present. A diagnosis of ATN in the presence of NSAID and dehydration was retained. One week after the initial evaluation, the proteinuria showed a complete regression and the renal function normalized within 3 weeks.
Case 3
The third patient was a 24-year-old female nurse. The patient presented to our outpatient clinic after a history of bilateral flank pain following moderate alcohol consumption and documented increased proteinuria on urine dipstick. On presentation, she was hemodynamically stable with persistent flank pain. She did not suffer from myalgia, arthralgia, fatigue, or headache. The remaining systemic history, medical history, and family history were unremarkable except for nicotine abuse. The clinical examination was unremarkable apart from kidney tenderness on percussion on both sides. Laboratory testing revealed an AKI I and proteinuria of 1.7 g/day at initial presentation. There was no evidence of urinary tract infection, leukozyturia or erythrocyturia. Eosinophilia was not assessed. In the medical history, the patient reported suffering from headache and taking ibuprofen on the day she consumed alcohol. The days before presentation were unusually hot weather and the patient did not follow adequate fluid intake. She does not use any other medication except for contraception.
On the following day, proteinuria already showed rapid regression to 500 mg/day and creatinine continued to increase. Because creatinine increased continuously during the first days, further diagnostics were performed although there was no clinical or laboratory evidence of vasculitis. Laboratory chemistry showed no evidence of ANCA vasculitis, hepatitis, or complement-mediated disease. Sonographically and on MRI, the kidneys showed borderline enlargement with preserved corticomedullary differentiation. A kidney stone or an obstructive of the kidneys were excluded.
In the context of a typical history of pain after alcohol consumption, NSAIDs and dehydration, the diagnosis of ATN was retained. Under conservative therapy with peroral fluid administration and analgesic therapy excluding NSAIDs, the renal values were completely regressed after 10 days, and the patient was symptom-free.
Discussion
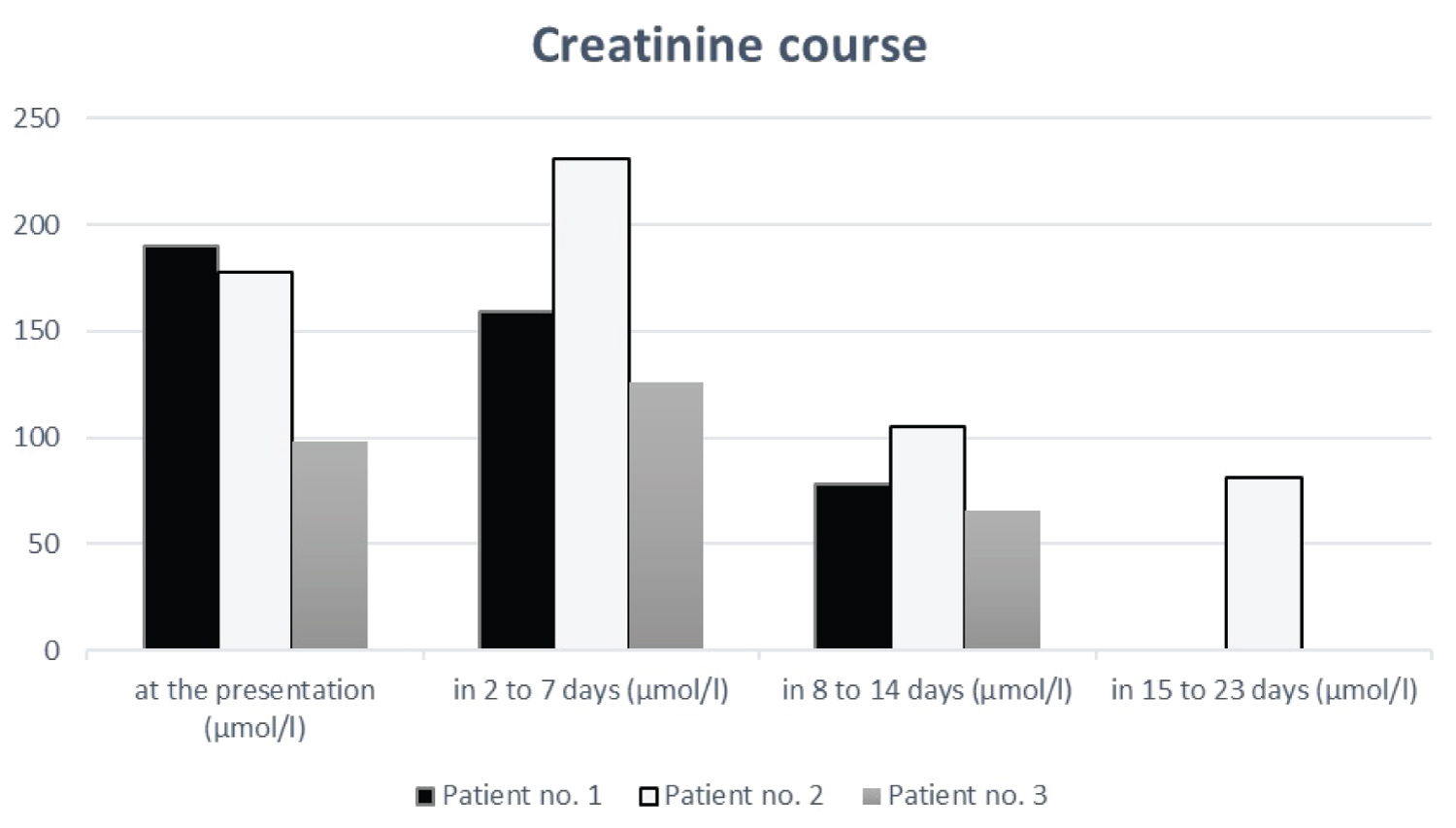
These three young patients have a similar history with ATN in the presence of NSAID and dehydration. Two of three patients were initially hospitalized with an incorrect diagnosis and received different medications and further diagnostics. Renal biopsy was avoided in all three cases because of rapid and complete remission with hydration and complete avoidance of NSAID. The kidney values recovered within 1-3 weeks as shown in Figure 1. The diagnosis of ATN in the presence of NSAID and dehydration is difficult based on laboratory findings. The medical history of the time before consultation focusing on NSAIDs consumptions and concomitant dehydration is important. A wrong diagnosis can lead to unnecessary kidney biopsy, unnecessary therapies and in the worst case to unnecessary interventions.
The main mechanism of NSAID action is the inhibition of the enzyme cyclooxygenase (COX). High doses of NSAIDs block both COX enzyme isoenzymes, COX-1 (cyclooxygenase-1) and COX-2 (cyclooxygenase-2) [2,3]. COX-1 is present in most cells and has regulatory and protective effects. COX-2 is activated by inflammation and proinflammatory cytokines [2].
NSAIDs can cause two different forms of acute kidney injury, hemodynamically mediated on the one side and acute interstitial nephritis on the other side [9]. The cornerstone of the pathogenesis of hemodynamically mediated AKI is inhibition of COX-1 enzymes with subsequent reduction of prostaglandin (PG) synthesis [10]. PGs do have no significant role in renal hemodynamics under normal physiological conditions [11]. In cases of sustained renal vasoconstriction, such as in patients with intravascular volume depletion or chronic kidney disease, PG-mediated afferent arteriolar vasodilation plays an important role in maintaining renal blood flow and glomerular filtration rate by reducing pre glomerular resistance [9,10]. Changes in renal medullary pO2 occur in human during water diuresis as evaluated by blood oxygenation level-dependent magnetic resonance imaging (BOLD MRI). Dehydration decreased renal medullary oxygenation in healthy volunteers. After water loading to induce water diuresis, renal medullary oxygenation levels improved significantly in all subjects. In the presence of cyclooxygenase inhibition by naproxen, the improvement in oxygenation during water diuresis was completely abolished [12,13]. This mechanism of the combined effect to decreased water diuresis (dehydration) and NSAIDs induced COX inhibition leading to decreased medullary oxygenation in human might well be the underlying mechanism of ATN in these three patients described above.
AIN is an inflammatory pathology of renal interstitium and tubules and occurs on allergic basis with edema, acute damage, and can potentially heal with interstitial fibrosis. This is not dose-dependent [11,14]. AIN occurs predominantly due to drugs such as beta-lactam antibiotics and sulfur-containing drugs [15,16]. The patients present with acute or subacute onset of allergic features such as rush, eosinophilia, and fever within a few days of starting the medication [15,16]. In recent years, other classes of drugs such the immune checkpoint inhibitors, proton pump inhibitors (PPI), NSAIDs, anticonvulsants, allopurinol, and some of the newer agents, such as the anti-angiogenesis agents also lead to AIN [1,11,15,17]. AIN triggered by these newer drugs does not have the same allergic features and have significantly longer latent periods as their predecessors. The classic triad of fever, rash and eosinophilia was present in only 10% of antibiotic-induced AIN cases [5,15].
The differentiation of ATN and AIN in patients with NSAID-related AKI in the context of dehydration is difficult. The clinical and laboratory signs are nonspecific [4]. In Table 1 we listed the possible small differences.
Patients, who develop ATN after exposure to NSAIDs often present with AKI, have minimal proteinuria and bland urine sediment. The development of AKI is often dose-dependent and depending on the triggering drug occurs earlier or later after exposure to an agent [14]. In the context of ATN, patients present with no evidence of system involvement and in some cases flank pain can be observed [17,18]. This was described as early as 1992 in two college students who developed acute tubular necrosis and flank pain after binge drinking beer and taking NSAIDs [18]. The kidneys of patients with ATN appear pale and swollen on imaging [14]. In the majority of cases, renal function fully recovers after discontinuation of the drug and initiation of supportive measures [14].
Crucial in the differential diagnosis of AIN and ATN is the medical history, the clinical evaluation of the patient and determination if a systemic process is present or if there was exposure to a suspected drug [17]. In the case of AIN, nonspecific symptoms such as general malaise, fatigue, weakness, and anorexia are observed. In addition, the patients describe myalgias, arthralgias, flank pain, normal blood pressure, no edema and fever [4,17,19,20]. Drug rash is reported in the literature in 15-50% of AIN cases [17].
Reliable and noninvasive diagnostic test for the clinical diagnosis of AIN and ATN are not available. Varieties of clinical tools are used by clinicians for diagnosis, including focused history, physical examination, imaging tests and certain laboratory data [15,17]. Eosinophilia may suggest an AIN-related immunoallergic disease, but eosinophilia may also occur in other clinical conditions, such as vasculitis, malignancy, and cholesterol embolism syndrome [19]. Previously, the urine eosinophil test was the main diagnostic test for AIN. A recent study showed that the sensitivity and specificity of the urine eosinophil test is low. Urinary eosinophils are seen not only in AIN but also in ATN, multiple myeloma, glomerulonephritis and atheroembolic disease [15]. Fogazzi BG, et al. found WBCs (white blood cells) and RBCs (red blood cells) most frequently in the urine of patients with AIN, but he also saw WBC casts, RTEC (renal tubular epithelial cell) and RTEC casts. Unexpectedly, the authors found RBC casts, which are so uncommon in AIN that some authors consider them a rule-out diagnosis for AIN [21].
Standard, rapidly available imaging of the kidneys such as ultrasound or CT scan is commonly used in patients with AKI. These modalities have their most important diagnostic role in excluding other causes of AKI [17].
Renal biopsies for histological diagnoses carry risks for patients and are not feasible in some individuals at increased risk of bleeding. As a result, delays in diagnosis and therapy may occur. Consequently, noninvasive biomarkers are need for the diagnosis of AIN [15]. The use of gallium-67 scintigraphy for diagnosis of AIN is not a universal practice and is controversial [22]. In 2019, Moledina and Parikh found higher urinary TNF-α (tumor necrosis factor alpha) and interleukin-9 in patients with biopsy-confirmed AIN than in ATN [15]. However, additional studies are need in this area.
NSAIDs are a known cause of AKI, but because renal failure is often mild, asymptomatic, transient, and nonanuric may be underdiagnosed [8,11].
Conclusion
OTC (over the counter) products are generally considered safe and are poorly monitored. NSAIDs pose an increased risk of renal injury but the incidence of AKI due to NSAIDs is not well established. In patients taking NSAIDs who have intravascular volume contraction, they pose a real risk for AKI. Clinicians must be cautious when treating patients with NSAIDs, not only in older patients and patients with preexisting renal disease, but also in younger patients at risk of intravascular volume depletion. Diagnosis of ATN in the presence of NSAID and dehydration is difficult. The medical history is important and can help us to establish the diagnosis.
Author Contributions
CC and BV designed the study; CC performed the literature search; CC, DS and BV wrote the paper.
References
- Musu M, Finco G, Antonucci R, et al. (2011) Acute nephrotoxicity of NSAID from the foetus to the adult. Eur Rev Med Pharmacol Sci 15: 1461-1472.
- Lucas GNC, Leitão ACC, Alencar RL, et al. (2019) Pathophysiological aspects of nephropathy caused by non-steroidal anti-inflammatory drugs. J Bras Nefrol 41: 124-130.
- Zhang J, Ding EL, Song Y (2006) Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: Meta-analysis of randomized trials. JAMA 296: 1619-1632.
- Clavé S, Rousset-Rouvière C, Daniel L, et al. (2019) The invisible threat of non-steroidal anti-inflammatory drugs for kidneys. Front Pediatr 7: 520.
- Nast CC (2017) Medication-induced interstitial nephritis in the 21st Century. Adv Chronic Kidney Dis 24: 72-79.
- Bindu S, Mazumder S, Bandyopadhyay U (2020) Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem Pharmacol 180: 114147.
- Zhang X, Donnan PT, Bell S, et al. (2017) Non-steroidal anti-inflammatory drug induced acute kidney injury in the community dwelling general population and people with chronic kidney disease: Systematic review and meta-analysis. BMC Nephrol 18: 256.
- Balestracci A, Ezquer M, Elmo ME, et al. (2015) Ibuprofen-associated acute kidney injury in dehydrated children with acute gastroenteritis. Pediatr Nephrol 30: 1873-1878.
- Ungprasert P, Cheungpasitporn W, Crowson CS, et al. (2015) Individual non-steroidal anti-inflammatory drugs and risk of acute kidney injury: A systematic review and meta-analysis of observational studies. Eur J Intern Med 26: 285-291.
- Misurac JM, Knoderer CA, Leiser JD, et al. (2013) Nonsteroidal anti-inflammatory drugs are an important cause of acute kidney injury in children. J Pediatr 162: 1153-1159.
- Dixit M, Doan T, Kirschner R, et al. (2010) Significant acute kidney injury due to non-steroidal anti-inflammatory drugs: Inpatient setting. Pharmaceuticals (Basel) 3: 1279-1285.
- Prasad PV, Epstein FH (1999) Changes in renal medullary pO2 during water diuresis as evaluated by blood oxygenation level-dependent magnetic resonance imaging: Effects of aging and cyclooxygenase inhibition. Kidney Int 55: 294-298.
- Tumkur SM, Vu AT, Li LP, et al. (2006) Evaluation of intra-renal oxygenation during water diuresis: A time-resolved study using BOLD MRI. Kidney Int 70: 139-143.
- Markowitz GS, Perazella MA (2005) Drug-induced renal failure: A focus on tubulointerstitial disease. Clin Chim Acta 351: 31-47.
- Moledina DG, Parikh CR (2019) Differentiating acute interstitial nephritis from acute tubular injury: A challenge for clinicians. Nephron 143: 211-216.
- Moledina DG, Wilson FP, Pober JS, et al. (2019) Urine TNF-a and IL-9 for clinical diagnosis of acute interstitial nephritis. JCI Insight 4: e127456.
- Perazella MA (2017) Clinical approach to diagnosing acute and chronic tubulointerstitial disease. Adv Chronic Kidney Dis 24: 57-63.
- Wen SF, Parthasarathy R, Iliopoulos O, et al. (1992) Acute renal failure following binge drinking and nonsteroidal antiinflammatory drugs. Am J Kidney Dis 20: 281-285.
- Caravaca-Fontán F, Fernández-Juárez G, Praga M (2019) Acute kidney injury in interstitial nephritis. Curr Opin Crit Care 25: 558-564.
- Rossert J (2001) Drug-induced acute interstitial nephritis. Kidney Int 60: 804-817.
- Fogazzi GB, Ferrari B, Garigali G, et al. (2012) Urinary sediment findings in acute interstitial nephritis. Am J Kidney Dis 60: 330-332.
- Graham F, Lord M, Froment D, et al. (2016) The use of gallium-67 scintigraphy in the diagnosis of acute interstitial nephritis. Clin Kidney J 9: 76-81.
- Joaquim AI, Mendes GE, Ribeiro PF, et al. (2010) Ga-67 scintigraphy in the differential diagnosis between acute interstitial nephritis and acute tubular necrosis: An experimental study. Nephrol Dial Transplant 25: 3277-3282.
- Hanif MO, Bali A, Ramphul K (2021) Acute renal tubular necrosis. StatPearls Publishing.
- Cavanaugh C, Perazella MA (2019) Urine sediment examination in the diagnosis and management of kidney disease: Core curriculum. Am J Kidney Dis 73: 258-272.
Corresponding Author
Cecilia Czerlau, MD, Department for Nephrology and Hypertension, University Hospital Insel Bern, Freiburgstrasse, CH-3010 Bern, Switzerland.
Copyright
© 2021 Czerlau C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.