Impact of Genetic Testing in the Diagnosis and Management of Sucrase-Isomaltase Deficiency
Abstract
The proband was of Arabic descent and evaluated for intractable diarrhea and poor growth. Molecular testing identified a nonsense mutation in the sucrase-isomaltase (SI) gene consistent with a diagnosis of sucrase-isomaltase deficiency (CSID). Appropriate treatment led to resolution of symptoms and improved growth. This case represents the first report of CSID with molecular confirmation in a patient without known European ancestry and demonstrates the importance of genetic testing for congenital diarrheal disorders.
Keywords
Congenital diarrheal disorder, Consanguinity, Sucrase-isomaltase deficiency
Introduction
Congenital diarrheal disorders (CDD) comprise a group of rare enteropathies that are typically monogenic and autosomal recessive manner in nature [1]. For many of these conditions, patients present within the first few days to weeks of life with severe diarrhea and dehydration. Congenital sucrase-isomaltase deficiency (CSID) is an autosomal recessive CDD, the result of mutation of the SI gene. This gene encodes a type II transmembrane glycoprotein of the intestinal brush border responsible for processing of dietary carbohydrates. Unlike many CDDs, however, patients with CSID often have delayed symptom onset. The classic presentation is that of an infant or child who develops severe watery diarrhea and failure to thrive following the introduction of sucrose or starch [2]. The disorder is commonly found in the circumpolar regions of Greenland and the Inuit populations of Alaska and Canada. It has an estimated incidence of 0.2-0.05% among individuals of European descent [3-6] and the vast majority of patients carry one of four abnormal alleles [7]. The most common of these alleles is a founder mutation with high prevalence among the Inuit people [8]. Additional mutations restricted to a few individuals or families have been documented [7] but are uncommon. Outside of these at risk populations, the condition is exceedingly rare.
Diagnosis of a congenital diarrheal illness is challenging and often complicated by heterogeneous etiology, overlap in phenotype, and a broad differential. Traditional approach relies heavily on defining the clinical characteristics of the illness and histological evaluation of intestinal tissue. Diagnostic yield of endoscopy, however, is highly variable. Increasingly, genetic testing has been implemented, facilitating rapid diagnosis and disease specific management. The advent of next gen (NGS) and whole exome sequencing has revolutionized the approach to this diverse group of disorders and proven an invaluable tool in identifying rare and novel syndromes. The utilization of molecular testing in conjunction with detailed phenotyping in CDD, however, has not yet become routine practice despite evidence that early molecular diagnosis has a direct impact on patient management [9].
Clinical Report
The proband was a 5-year-old female referred from the United Arab Emirates for evaluation of failure to thrive and chronic diarrhea. Onset of symptoms was reported to be 4 months of age, coincident with the introduction of soft foods to her diet. Stools were described as frequent and watery with as many as twelve per day.
Indirect pancreatic function testing (PFT) was done by measuring the zymogen elastase-1 in stool. This was initially performed by ARUP laboratories, Salt Lake City, UT. Results were suggestive of possible exocrine pancreatic insufficiency (107 μg/g). This was repeated again two months later as she remained symptomatic. Stool sample was analyzed at Kaleida Health Children's Hospital laboratory, Buffalo, NY, using an enzyme linked immunosorbent assay (ELISA) manufactured by Bioserv Diagnostic GmbH, Germany. Repeat results again showed low fecal elastase (101.8 μg/g of stool). A trial of pancreatic enzyme supplementation therapy was started, however, symptoms persisted.
Direct PFT was done endoscopically with a pre-specified protocol, which involved collection of secretions at set intervals from the second portion of the duodenum, close to the ampulla of Vater, after pancreatic stimulation with Sinaclide (synthetic C-terminal octapeptide of cholecystokinin). For this patient, three aliquots were collected with confirmed pH between 7.5 and 7.7. Samples were sent to Kalieda Health Children's Hospital Laboratory, Buffalo, NY, for measurement of trypsin, amylase, lipase, chymotrypsin and protein. All pancreatic enzyme activities were reported as normal therefore diagnostic workup was expanded.
Intermittent supplementation with pancreatic enzymes over a period of several years resulted in minimal improvement in symptoms. Family reported normal appetite and caloric intake, but weight was less than 3rd centile for age. The decision was made to proceed with esophagastroduodenoscopy and colonoscopy for further evaluation. Duodenal mucosa was noted to be friable and the ampulla appeared umbilicated and edematous on gross examination, however, no diagnostic abnormalities were visible on histological examination. Tissue was collected for disaccharidase analysis, which was sent to Kaleida Health Children's Hospital Laboratory, Buffalo, NY. Dissachariadases were analyzed by the Dahlqvist method [10]. Residual sucrase and palatinase (isomaltase analog) activity was 0 μM/min/g protein consistent with a diagnosis of sucrase-isomaltase deficiency (Table 1).
Genetic Testing
Family history was significant for consanguinity, the proband was the product of a first cousin union. Microarray demonstrated large tracts of homozygosity encompassing 6.5% of the genome with increased risk for unmasking of recessive conditions. Candidate genes within the homozygous tracts and consistent with the patient's phenotype included the SLC26A3 gene for congenital chloride diarrhea, and the SI gene associated with CSID.
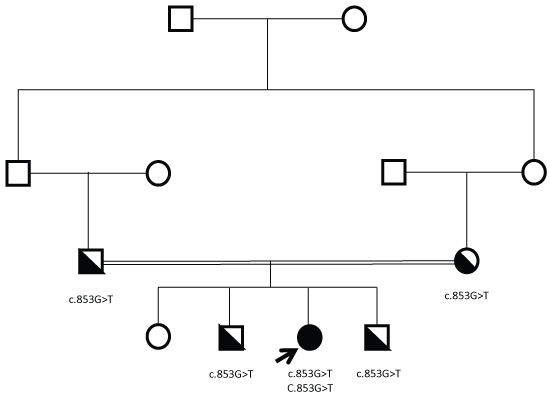
Genomic DNA was isolated from the proband and the entire coding region as well as all exon/intron boundaries of the SI gene was analyzed by PCR and bi-directional sequencing. A novel nonsense mutation, c.853G > T (p.E285*), was identified. This change results in a premature stop codon and substantially truncated mutant protein length [11]. Subsequent genetic testing in first-degree family members demonstrated heterozygous state in the mother, father, and two siblings (Figure 1).
Treatment
Pancreatic enzyme supplementation was discontinued and patient was instructed to begin sucrosidase with snacks and meals in addition to restriction of complex carbohydrates and sucrose containing foods. Weight gain improved with return of normal linear growth. Body mass index increased to the 26th centile during the six month follow up period and symptoms were largely ameliorated except when patient deviated from recommended diet.
Discussion
The non-specific presentation of congenital diarrheal illnesses makes diagnosis challenging, however, rapid identification of the disorder can have significant impact on management and outcome. Over 50 genes have been implicated in this group of disorders with substantial overlap in phenotype [9,12]. In this patient, genetic diagnosis helped guide targeted intervention with resolution of symptoms and improved growth. This type of molecular approach is not limited to CSID and rapid identification of the disorder can have significant impact on management and outcome. Genotype specific therapies are increasingly utilized for many congenital diarrheal illnesses. Hematopetic stem cell transplant is potentially curative in disorders such as IL10 signaling defects [13], XIAP deficiency [14], and FOXP3 deficiency [15]. Early recognition of congenital chloride diarrhea, associated with mutations in SLC26A3 has been proven to mitigate renal complications and restore normal growth and development [16]. Even in conditions for which there is no ameliorative treatment, such as trico-hepato-enteric syndrome (SKIV2L and TTC37), early confirmation of diagnosis via genetic testing and implementation of long term TPN can improve quality of life and reduce associated comorbidities [17].
Increasingly, international families are seeking care within the United States. Among residents of North Africa, the Middle East, and West Asia, first cousin unions are common and comprise an estimated 20-30% of all marriages [18]. In populations where consanguinity is practiced, however, there is higher risk for rare, autosomal recessive conditions to occur. Although microarray cannot identify single gene changes, it can be used to isolate homozygous tracts within the genome that result from consanguinity. These tracts can then be interrogated for candidate genes consistent with the patient's phenotype, as was done in this case, eliminating the need for broader genetic testing.
Molecular testing was recently been proposed as a first line tool for diagnosis and management of CSID, a safer alternative to sedation and traditional endoscopy. A study by Uhrich [7] confirmed that 83% of symptomatic children with clinical suspicion for CSID and European ancestry could be diagnosed via genetic testing alone. Molecular testing should be considered as a first line diagnostic tool and may be more appropriate than traditional endoscopy in clinically tenuous patients.
Although fecal elastase studies in this patient were suggestive of possible exocrine pancreatic insufficiency, intermittent supplementation with pancreatic enzymes over a period of several years resulted in minimal improvement in symptoms. Fecal elastase is an indirect measure of pancreatic function and can be falsely positive in the setting of diarrhea, exemplified by this case. Direct pancreatic function testing was normal and the patient has had improved linear growth and weight gain with resolution of her diarrhea in the absence of pancreatic enzyme supplementation and strict adherence to her diet.
This is the first report of a patient with CSID and confirmed mutation in the SI gene without known European or Inuit ancestry. At this time, there are no reports of patients of Arabic or African descent with CSID reported in the literature. Geng, et al. [2] described three Chinese patients with a phenotype consistent with CSID however; neither sucrase-isomaltase activity nor molecular sequencing was performed. In the absence of additional data, it is difficult to estimate the actual prevalence of the disorder in their target population. It is possible that given its rarity, CSID is excluded as a potential etiology and therefore patients traditionally believed to be not at risk escape diagnosis.
This case serves to emphasize that rare and uncommon disorders should be equally considered if consistent with the phenotype. Given high diagnostic yield in CSID, molecular testing should be considered as a first line tool as rapid identification and implementation of appropriate therapy results in resolution of symptoms.
Conflicts of Interest
There are no conflicts of interest to disclose. No monetary support was received for this publication.
References
- Canani RB, G Terrin (2011) Recent progress in congenital diarrheal disorders. Current Gastroenterology Reports 13: 257-264.
- Geng L, Li DY, Ou W, et al. (2014) Congenital sucrase-isomaltase deficiency: an under-diagnosed disease in Chinese children. BMC Pediatr 14: 11.
- Bell RR, Draper HH, Bergan JG (1973) Sucrose, lactose, and glucose tolerance in northern Alaskan Eskimos. Am J Clin Nutr 26: 1185-1190.
- Ellestad-Sayed J, Haworth JC, Hildes JA (1978) Disaccharide malabsorption and dietary patterns in two Canadian Eskimo communities. Am J Clin Nutr 31: 1473-1478.
- Peterson M, Herber R (1967) Intestinal sucrase deficiency. Trans Assoc Am Physicians 80: 275-283.
- Welsh JD, Poley JR, Bhatia M, et al. (1978) Intestinal disaccharidase activities in relation to age, race, and mucosal damage. Gastroenterology 75: 847-855.
- Uhrich S, Wu Z, Huang JY, et al. (2012) Four mutations in the SI gene are responsible for the majority of clinical symptoms of CSID. J Pediatr Gastroenterol Nutr 55: S34-S35.
- Marcadier JL, Boland M, Scott CR, et al. (2015) Congenital sucrose-isomaltase deficiency: identification of a common Inuit founder mutation. CMAJ 187: 102-107.
- Kammermeier J, Drury S, James CT, et al. (2014) Targeted gene panel sequencing in children with very early onset inflammatory bowel disease-evaluation and prospective analysis. J Med Genet 51: 748-755.
- Dahlqvist A (1964) Method for assay of intestinal disaccharidases. Anal Biochem 7: 18-25.
- Schwarz JM, Cooper DN, Schuelke M, et al. (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11: 361-362.
- Canani RB, Castaldo G, Bacchetta R, et al. (2015) Congenital diarrhoeal disorders: advances in this evolving web of inherited enteropathies. Nat Rev Gastroenterol Hepatol 12: 293-302.
- Engelhardt KR, Shah N, Faizura-Yeop I, et al. (2013) Clinical outcome in IL-10-and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J Allergy Clin Immunol 131: 825-830.
- Marsh RA, Rao K, Satwani P, et al. (2013) Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood 121: 877-883.
- Barzaghi F, Passerini L, Bacchetta R (2012) Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol 3: 211.
- Lechner S, Ruemmele, Frank M, et al. (2011) Significance of molecular testing for congenital chloride diarrhea. Journal of Pediatric Gastroenterology and Nutrition 53: 48-54.
- Fabre A, Martinez-Vinson C, Goulet O, et al. (2013) Syndromic diarrhea/Tricho-hepato-enteric syndrome. Orphanet J Rare Dis 8: 5.
- Bittles A (2008) A community genetics perspective on consanguineous marriage. Community Genet 11: 324-330.
Corresponding Author
Dr. Rachel C Lombardo, Department of Human Genetics, Cincinnati Children's Hospital and Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229, USA, Tel: +513-636-4760, Fax: +513-636-7297.
Copyright
© 2017 Lombardo RC, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.