GNUMAP 4.0: Space and Time Efficient NGS Read Mapping Using the FM-Index
Abstract
In this article, we present GNUMAP (Genomic Next-generation Universal MAPper), a next-generation sequence read mapper, that utilizes a Full-text index in Minute Space (FM-index) with the Burrows-Wheeler Transform (BWT) as the reference genome data structure. Using the FM-index, GNUMAP is able to map reads with a dramatic decrease in memory usage while maintaining the same probabilistic mapping characteristics of previous versions. GNUMAP is now able to map Next-generation sequencing reads using a variable kmer size without having to rebuild the reference genome index. Other enhancements were also made to reduce the number of costly Needleman-Wunsch alignments during mapping. This article shows the general benefits of using the FM-index as a reference genome index data structure versus GNUMAP's previous data structure, the hashmap. Additionally, we show that while the FM-index is sometimes slower than the hashmap, parameter tuning increases mapping speeds significantly, uses less memory and maintains the probabilistic read mapping characteristics when compared to the hashmap. The new version of GNUMAP is available at https://github.com/byucsl/gnumap
Index Terms
FM-index, NGS, NGS read mapping
Background
Next-generation sequencing (NGS) read mapping continues to be an important process in many "-omic" analysis pipelines. The ability to map NGS reads with speed and accuracy has been addressed by many algorithms including Bowtie2 [1], BWA [2], GNUMAP [3] and SOAP [4]. The Genomic Next-generation Universal MAPper (GNUMAP) is a mapping algorithm that differentiates itself from other mappers by using a probabilistic Needleman-Wunsch (NW) alignment algorithm as well as calculating a posterior probability score for multi-mapped reads [3]. The probabilistic NW is unique because it is able to probabilistically align reads to the reference genome by using the raw Solexa/Illumina intensity or probability files. This results in increased confidence in mapping because all possible bases at a particular position are aligned with base-specific uncertainty taken into account. The probabilistic read scoring approach used by GNUMAP is designed to map reads from repeat regions and reduce discarded reads. While the probabilistic mapping processes in GNUMAP has benefits, GNUMAP has remained difficult to use. The data structure used to store the indexed reference genome used for mapping can be too large to fit into memory on many machines, requiring nearly 40GB RAM for the human reference genome for certain kmer sizes.
GNUMAP previously relied on a hashmap to index a reference genome similar to other NGS read mappers such as SeqMap [5], RMAP [6], and ELAND (Cox, unpublished software). We denote GNUMAP using the hashmap data structure as GNUMAP-hashmap. The hashmap is built by using the kmer as the key in the map. The value for a particular key consists of the genomic coordinates of all occurrences of that kmer in the reference genome. As the hashmap is being built, a maximum threshold (the maximum kmer occurrence) is used to remove kmers that occur too frequently in the genome. This reduces the size of the index and speeds up mapping as kmers that occur more frequently throughout the genome cause frequent false-positive mapping attempts.
The hashmap data structure provides the benefit of quickly looking up genomic coordinates for kmers. It is, however, also limited in several ways. The main limitations are the amount of memory required to hold the entire hashmap in memory and the inability to reuse the hashmap for different length kmers. The hashmap could become extremely large when used with long kmers on a large reference genome. It also needs to be rebuilt for different sized kmers as the hashmap is built with a specific and immutable kmer size. The hash is also limited because it cannot be constructed for kmers where k > 15. This limits the usable kmer sizes that can be used during mapping. A variant of GNUMAP uses the C++ Standard Template Library (GNUMAP-STL) to hash the genome to overcome the maximum kmer size limit. To overcome these weaknesses and retain the strengths afforded by probabilistic read mapping, GNUMAP has been modified to make use of an Full-text index in Minute space (FM-index) as the data structure used for indexing a reference genome.
The FM-index is a text compression algorithm that allows for fast substring look-ups in the indexed text [7]. There are many mappers that exist that use this same data structure such as BWA [2]. In this work, we show the effects of implementing the FM-index from BWA in GNUMAP (referenced as GNUMAP-FM). We will show that run-time only suffers moderately while the amount of memory used is greatly reduced allowing GNUMAP to run on commodity desktop hardware with arbitrary kmer sizes. Using the FM-index, GNUMAP is also enhanced by not needing to create multiple reference genome indices for different length kmers. The FM-index also allows for kmers of arbitrary length to be searched for, thus overcoming the kmer size limit imposed by the hash data structure. While changing the underlying data structure used for fast kmer look-ups for read mapping, GNUMAP is still able to maintain the same mapping probabilistic properties from previous versions.
In this article, we discuss the improvements implemented in GNUMAP and compare the new version of GNUMAP to the previous version. First, we describe the FM-index Implementation and evaluation datasets used, then we benchmark the new version compared to the old version based on run-time and amount of RAM used. Finally, we discuss the flexibility of parameter tuning made possible by the new version as well as future work.
Methods
Enhancements to GNUMAP include: The implementation of an FM-index for reference genome indexing, added parameters for mappings based solely on kmer look-ups, reduced NW alignments by removing alignment re-computations and other bug fixes. The FM-index and other optimizations were made to GNUMAP version 3.0.2. We release this update as GNUMAP version 4.0.
FM-Index implementation
The FM-index as implemented in BWA [8] is version 2 of the FM-index using the Burrows-Wheeler transform, which we used as a drop-in replacement for the hashmap used originally in GNUMAP. The FM-index is a succinct data structure that compresses the genome and allows fast (sub-linear) substring searches. Users should note that the FM-index in BWA replaces ambiguous bases in the reference sequence randomly with an A, C, G or T. By replacing ambiguous characters, the alphabet of the indexed genome is reduced to four characters and enables 2-bit encoding of the genome that is more space efficient than using 3-bits or storing the genome as plain text. This randomness added during reference genome indexing causes GNUMAP to produce different results when using the same parameters between the FM-index and hashmap versions. While this is problematic, we find that that these issues are only problematic when using small kmers (k < 15) because the shorter the kmer the more likely that a particular kmer is randomly found after the ambiguous base substitution. When using larger kmers (k ≥ 15) and additional parameter tuning we see the mapping run-times decrease and number of reads mapped increase drastically (see Table 1 for benchmarks).
Additional optimizations and bug fixes
In addition to using the FM-index, two other modifications and other bug fixes were made to GNUMAP. First, we implemented a method for mapping reads without using the probabilistic Needleman-Wunsch that is based solely on genomic kmer location look-ups. Second, we modified the mapping algorithm to reduce the number of NW alignments that occur during mapping. Performing the NW alignment is the most costly operation done in GNUMAP. We found that previous versions of GNUMAP would often recompute alignments it had already done. Fixing this resulted in much fewer alignments and much quicker run-times. These modifications, bug fixes and the indexing behavior of the BWA FM-index cause mapping results to vary between the current and previous version of GNUMAP.
Datasets
We compared the versions of GNUMAP on how quickly the reads were aligned, how much memory was used to align the reads, and how accurate the alignment was. Four different datasets were used, three simulated and one real dataset.
WgSim dataset
A smaller simulated dataset was generated using WgSim [9]. This smaller dataset consists of 50,000 synthetic 100 bp NGS reads from the human reference genome hg19 and was used exclusively for memory profiling benchmarks.
ART parameter tuning
This dataset consists of 28,880 synthetic 100 bp reads generated from the human reference genome hg19 using ART [10]. Using this dataset, we tested the mapping performance of GNUMAP with the FM-index. This dataset was generated to ensure we have reads sampled uniformly across the whole genome to see general mapping performance.
ART mapping performance
We used the read simulation program ART [10] to generate synthetic 100 bp NGS single-end reads. ART generated 14,486,006 reads with 0.5× coverage of the hg19 human genome, with a 0.009% insertion rate and 0.023% deletion rate.
Real data
For the real data analysis, we used a set of NGS reads from the 1000 Genomes Project [11]. We downloaded the low coverage whole genome sequence reads (ERR251013), 65,820,186 total, from The International Genome Sample Resource (IGSR) for the individual HG00140.
All datasets were aligned to the hg19 human genome assembly. Furthermore, all experiments were run on the Fulton Supercomputing Laboratory's supercomputer, Mary Lou, using 12-core Intel Haswell (2.3 GHz) processors with 64GB of 2133 Mhz DDR4 RAM.
Benchmarking
Measuring memory usage
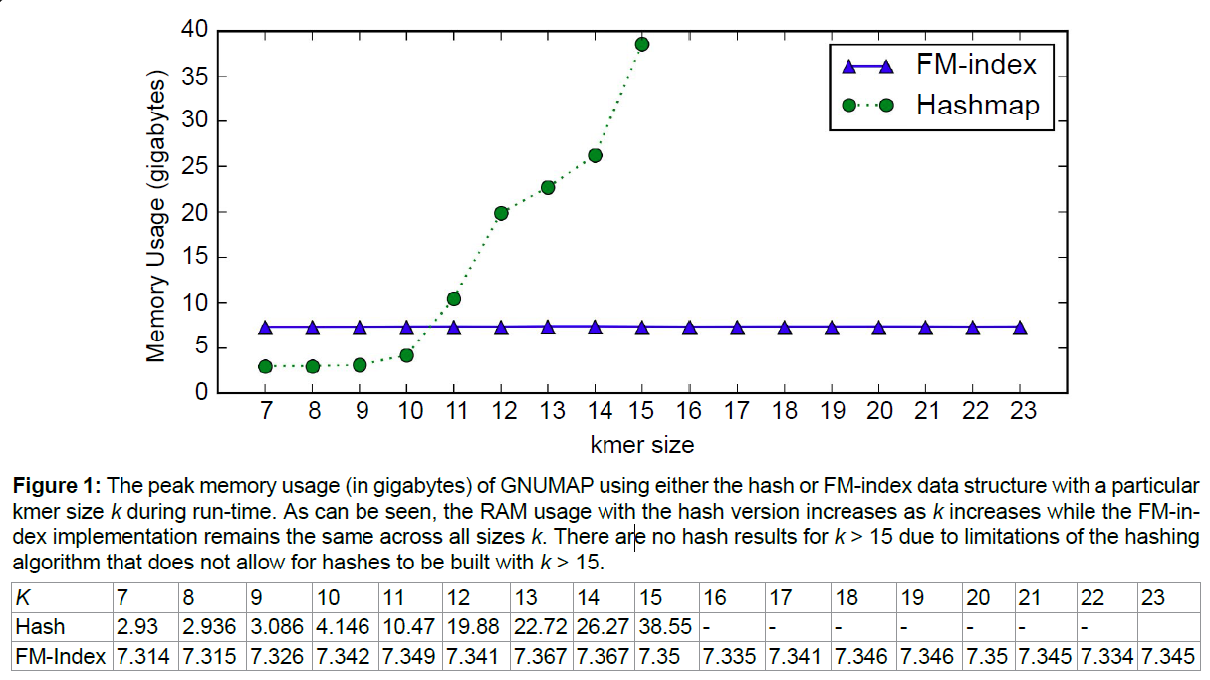
GNUMAP's memory consumption using the hash and the FM-index was profiled using Valgrind's heap profiler tool Massif [12]. Using Massif causes the profiled program to slow down tremendously. To profile memory usage, we used the smaller WgSim simulated dataset mapped to the human reference genome hg19. This allowed for memory profiling in a reasonable amount of time (Figure 1).
Parameter tuning
Parameter tuning with the FM-index shows that results as good, if not better, than GNUMAP-hashmap are possible. We not only measured the mapping time and percent reads mapped, but also how accurate the mappings were using the synthetic ART Parameter Tuning dataset.
Mapping results were verified against the ground truth during parameter tuning. We distinguish two types of mapping accuracy: All read accuracy and mapped read accuracy. All read accuracy considers all reads in the readset (mapped and unmapped). Only reads that mapped uniquely and to the correct location were counted as correct. Reads mapped to multiple locations (multi-mapped read) were counted as mapping incorrectly as well as any read that was left unmapped.
Mapped read accuracy is where we look only at mapped reads in the readset. Any read that is unmapped is not counted against the accuracy. Here also, only reads that map uniquely to the correct location are counted as correct. Mapped reads that do not map to the correct location or are multi-mapped reads are counted as incorrect.
Results and Discussion
The main metrics used to test the performance differences between the hash and FM-index versions of GNUMAP were mapping time and memory usage during mapping. Results were generated using various kmer sizes. For the FM-index, we tested using kmer sizes 7 ≤ k ≤ 23. We used the original GNUMAP hashing algorithm (GNUMAP-hashmap) for kmer sizes 7 ≤ k ≤ 15 and attempted to generate hashmap indices for k > 15. All plots were generated using matplotlib [13].
Memory usage
Memory usage during run-time was measured and can be seen in Figure 1. All parameters were maintained between the hashmap and the FM-index runs. Memory usage remains constant with all kmer sizes for the FM-index while the hashmap memory usage grows dramatically. Indexing a genome using the hashmap algorithm and k > 15 is not possible unless using the STL variant of GNUMAP.
GNUMAP-FM consumes a great deal less memory when running than the hashmap version. The hashmap index memory usage explodes in size as kmer size grows. The amount of memory required for the hashmap to run using a hash of the human reference genome and k = 15 is nearly 40 GB. We were unable to generate hashes with k > 15. This limitation poses a problem for read mapping because GNUMAP is unable to leverage longer kmers that increase read mapping speed. Because the FM-index uses a constant amount of memory at any size k any kmer size is a usable mapping parameter that can be tuned to maximize the number of mapped reads. The amount of memory used by the FM-index version allows GNUMAP to be run on even commodity desktop computers with all parameters available where the hashmap version is only able to run on higher-end machines with vast amounts of RAM.
Run-Time
A comparison of GNUMAP-hashmap to GNUMAP-FM measuring run-time differences can be seen in Table 2. Using the same parameters, GNUMAP-FM is able to outperform the hashmap version.
Despite having a slower indexing data structure, removing duplicate Needleman-Wunsch alignments provides significant speedups. The speedup due to alignment deduplication grows as the dataset becomes larger. This is because GNUMAP spends most of its time performing NW alignments and with more reads there are more alignments that are erroneously recomputed.
Mapping runs on the ERR251013 dataset for kmer sizes 11 and 12 did not finish within the given walltime (24 hours). On the ART synthetic dataset, the run-time for the 11 and 12-mer runs are much higher than those of other kmer sizes. This indicates that there is some characteristic of the kmer composition of hg19 at these particular sizes that made mapping difficult.
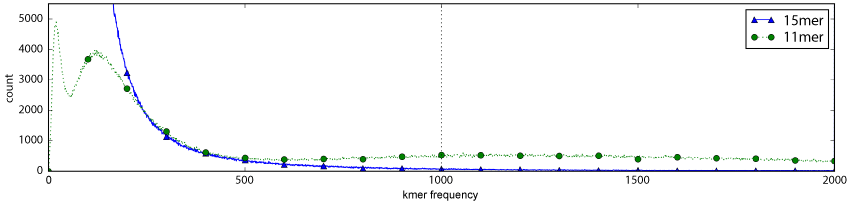
We generated kmer frequency profile plots shown in Figure 2 to understand why mapping is so much slower using 11 and 12-mers versus higher kmer sizes. Kmer counting and histogram counts were generated with Jellyfish [14]. We compared the distribution of the 11-mer (problematic size) with 15-mer (usable size). Kmer sizes smaller than 11 were not considered because their mapping performance is poor (see Table 2) and moving from 11-mers to 15-mers provides a large decrease (nearly 9 hours in the ART synthetic dataset) in run-time. The x-axis of Figure 2 is the kmer frequency, how often a particular kmer occurs in the genome. Moving from left to right on the x-axis corresponds to an increase in number of possible alignment operations. This is because kmers that occur more frequently in the genome result in more locations that GNUMAP must check for possible mapping. Kmers that have lower frequencies (left side of the plot) are better for mapping because they occur in fewer places in the human genome (more unique) and cause fewer alignment operations to occur. Alignments performed at genomic positions triggered by frequently occurring kmers are often false-positive mappings and usually discarded. Despite being discarded, the triggered alignment increases the run-time of GNUMAP because the alignment must be performed and scored to decide whether or not to discard it. Figure 2 shows the maximum kmer occurrence threshold at 1,000. The area between the 11-mer and 15-mer plots signifies why using an index with k = 11 is so much slower, there are many more 11-mers that have 500 ≤ frequency ≤ 1000 than 15-mers. This means that when 11-mers are used for indexing hg19, there are significantly more kmers that trigger more costly alignments in GNUMAP and result in a huge slowdown.
Looking at run-time and memory usage together reveal even greater limitations of GNUMAP-hashmap. Table 2 shows that execution times of using smaller kmers (7 ≤ k ≤ 10) result in very fast run-times and consume very little memory (< 3 GB) but very few reads mapped. When using 13 ≤ k ≤ 15, GNUMAP-hashmap runs very quickly and is able to map the majority of reads (especially in the ERR251013 dataset) but it uses enough memory to preclude some users from running it.
Index size on disk
GNUMAP-hashmap requires a new index to be built for every kmer size and maximum kmer occurrence. Using a different kmer size k results in different kmer profiles and affects the size of the hashmap that is generated. The sizes of the different hashes stored on disk for the human reference hg19 can be seen in Table 3. Going from k = 10 to k = 11 results in the hash doubling in size. This also occurs when going from k = 11 to k = 12. In general, the hash size is expected to roughly double every time kmer length is incremented. The hash, however, only stores kmers that occur within the genome resulting in hashes that don't double in size at each increment of k. The size of the index will also vary based on the maximum kmer occurrence threshold and the specific genome being indexed. GNUMAP-hashmap was unable to generate indices with k > 15, GNUMAP-STL can theoretically do so but was unable to for the human reference genome on a system with 128GB RAM. The size of the FM-index is 3.13GB on disk and only needs to be stored once for all sizes k. Storing a different hashmap to index the same information with different parameters is unnecessary when data structures like the FM-index exist.
Parameter tuning
Parameter tuning results are shown in Table 1. The Max Kmer Occ parameter is the threshold for the maximum number of times a kmer can occur in the genome to be used for mapping. The Min Kmer Hits parameter refers to the minimum number of kmer hits a read needs to trigger an NW alignment and be considered for mapping. Kmer Jump Size specifies the minimum distance between kmer hits in a read. Setting the kmer jump size to 1 results in each kmer from a read being looked up for mapping. Changing various parameters in GNUMAP-FM allows for as many, if not more, reads to be mapped as GNUMAP-hashmap in significantly less time.
Using the FM-index also allows for a great amount of flexibility that was not present when using the hashmap. When using the hashmap version, creating the genome index committed the user to the specific parameters used to build the index, there was no flexibility after it had been built and required a full rebuild if kmer size or maximum kmer occurrence were to be respecified.
Using the FM-index allows us to change these parameters to suit our needs from one sequencing run to another as the genomic composition of different samples can be different, thus requiring different parameters for optimal mapping. Tables 2 and the first two rows of 2 show that using the FM-index can cause a dramatic increase in run-time (nearly 4× longer) when identical parameters are used. However, by being able to change the size of k and the maximum kmer occurrence parameters (the two parameters that are immutable when initially creating the hashmap) we find that we can map more reads than the hashmap version in significantly reduced time. While using the FM-index with the same parameters causes a near 4× slowdown in runtime when compared to the hashmap, tuning the parameters causes a near 4× speedup while mapping more reads with higher all read accuracy and comparable mapped read accuracy. This huge speedup was afforded by our ability to choose a kmer size that is impossible when using either hashmap versions on a genome as large as the human genome. Specifically, setting k = 23 and the max kmer occurrence to 10, we were able to map more reads in less than 15 seconds versus the hashmap run which took nearly a minute.
Benefits of parameter tuning
Being able to dynamically change the maximum kmer occurrence also allows us to loosen or tighten our mapping criteria. Using a lower threshold allows for rarer kmers to indicate where we should map reads and decreases extraneous alignments that occur when a common kmer is used for mapping. With the same parameters, the FM-index had a significant slowdown and slight decreases in mapped read accuracy where a higher percentage of mapped reads from the hashmap mapped correctly compared to the FM-index. Using the flexibility of the FM-index, we find that the slight decreases in mapped read accuracy are offset because we're able to map more of the total readset resulting in an increased overall all read accuracy. We find that through this process we're not only able to map more reads about as accurately as the hashmap but we can do so in less time using less memory.
The benefits of flexible parameter tuning generalize even further when considering other reference genomes and different quality NGS reads. We found that setting a maximum kmer occurrence of 1,000 was not necessarily the best parameter when mapping to hg19. GNUMAP-hashmap was fixed to its initial parameter choices unless we decided to regenerate the entire hashmap. GNUMAP-FM could change all parameters on demand. When indexing other genomes, it may become apparent that when mapping reads that a higher or lower threshold for maximum kmer occurrence is necessary. This could be the case if using a genome that has many kmers that repeat throughout the genome with relatively fewer unique kmers.
Changing the size of the kmer is also very powerful especially when dealing with NGS reads of lower quality. This becomes extremely pertinent as new sequencing technologies, such as PacBio, generate extremely long reads (> 500 bp) and have error rates > 10% that are distributed relatively evenly throughout the read when compared to technologies like Illumina where errors are more likely to occur at the end of the read [15-17]. Mapping reads with error profiles similar to PacBio can necessitate using shorter kmers and increasing the maximum kmer occurrence threshold because shorter kmers are more likely to occur frequently throughout a genome than longer kmers.
Changing kmer size and max kmer occurrence thresholds is also beneficial when mapping reads from a specimen that is divergent from the reference genome being used. In studies where the reference genome is of low quality, as is the case for many non-model organisms, lowering kmer size and increasing the occurrence threshold allows for more ambiguous mapping positions to be evaluated. Source specimens can also be different from the reference in the case of highly polymorphic organisms or where high rates of heterozygosity are found and can affect general analysis and sequence processing [18]. The flexibility offered by GNUMAP-FM allows users to work much more easily with non-model organisms, highly polymorphic species and species with high rates of heterozygosity.
Conclusion
We have shown the benefits of using the FM-index in place of the hashmap data structure in GNUMAP. Initially, we were concerned about using the FM-index because the data structure is slower than the hashmap. Using the same parameters in GNUMAP-hashmap and GNUMAP-FM, we saw that it can result in a large slowdown in GNUMAP-FM's run-time. We find, however, that using parameter tuning coupled with the other optimizations and bug fixes overcomes the possible run-time issues. These results show the impact that parameter tuning has and how important it is to work with the composition of a specific dataset. Finding optimal mapping parameters automatically would be ideal but must take into account the kmer composition of the indexed reference genome and the type of DNA sequencing used. GNUMAP-hashmap is unable to change parameters without triggering a complete index rebuild while GNUMAP-FM, on the other hand, is able to switch between parameters extremely easily. We are now also able to use kmer sizes that were unavailable to us in either GNUMAP-hashmap or GNUMAP-STL. Our new release of GNUMAP-FM is an improvement over the previous version because it is able to map more reads in less time using less memory with about the same accuracy as well as adding flexibility to the user.
Acknowledgment
The authors would like to acknowledge the support of Dr. Christophe Giraud-Carrier, Nozomu Okuda and the members of the Computational Science Laboratory for their help and support during this project. We would also like to thank the Fulton Supercomputing Laboratory for their support of the hardware on which we ran these experiments.
References
- B Langmead, SL Salzberg (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9: 357-359.
- H Li, R Durbin (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25: 1754-1760.
- NL Clement, Q Snell, MJ Clement, et al. (2010) The GNUMAP algorithm: Unbiased probabilistic mapping of oligonucleotides from next-generation sequencing. Bioinformatics 26: 38-45.
- R Li, Y Li, K Kristiansen, et al. (2008) SOAP: Short oligonucleotide alignment program. Bioinformatics 24: 713-714.
- H Jiang, WH Wong (2008) SeqMap: Mapping massive amount of oligonucleotides to the genome. Bioinformatics 24: 2395-2396.
- D Smith, Z Xuan, MQ Zhang (2008) Using quality scores and longer reads improves accuracy of solexa read mapping. BMC Bioinformatics 9: 128.
- P Ferragina, G Manzini (2005) Indexing compressed text. Journal of the ACM 52: 552-581.
- M Burrows, DJ Wheeler (1994) A block-sorting lossless data compression algorithm. SRC Research Report.
- H Li (2011) Wg sim-read simulator for next generation sequencing.
- W Huang, L Li, JR Myers, et al. (2012) ART: A next-generation sequencing read simulator. Bioinformatics 28: 593-594.
- 1000 Genomes Project Consortium, Auton A, Brooks LD, et al. (2015) A global reference for human genetic variation. Nature 526: 68-74.
- N Nethercote, R Walsh, J Fitzhardinge (2006) Building workload characterization tools with valgrind. IEEE International Symposium on Workload Characterization.
- JD Hunter (2007) Matplotlib: A 2d graphics environment. Computing in Science & Engineering 9: 90-95.
- G Marcais, C Kingsford (2011) A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27: 764-770.
- MA Quail, M Smith, P Coupland, et al. (2012) A tale of three next generation sequencing platforms: Comparison of ion torrent, pacific biosciences and illumine MiSeq sequencers. BMC Genomics 13: 341.
- MO Carneiro, C Russ, MG Ross, et al. (2012) Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics 13: 375.
- K Nakamura, T Oshima, T Morimoto, et al. (2011) Sequence-specific error profile of illumina sequencers. Nucleic Acids Res 39: e90.
- MS Fujimoto, PM Bodily, N Okuda, et al. (2014) Effects of error-correction of heterozygous next-generation sequencing data. BMC Bioinformatics 15: S3.
Corresponding Author
Mark J Clement, Department of Computer Science, Brigham Young University, Provo, UT, USA.
Copyright
© 2018 Fujimoto MS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.