Stability-Indicating HPLC-DAD Determination of Lamivudine in Pharmaceutical Preparations and Biological Fluids
Abstract
A simple, selective, and stability-indicating high pressure liquid chromatographic method with diode array detection was developed for the analysis of lamivudine in tablets. The separation was performed on a C18 (octylsilyl) BDS Hypersil®column (4.6 × 150 mm × 5 μm), with isocratic elution of the mobile phase composed of acetonitrile and 50 mM phosphate buffer of pH 4 (adjusted using phosphoric acid) in a ratio of (10:90) v/v. The mobile phase was pumped at a flow rate of 1.0 mL/min. The detector was set at 280 nm and quantification of the analyte was based on peak area measurement. The method was validated with respect to linearity, range, precision, accuracy, selectivity, robustness, LOD and LOQ. The investigated drug exhibited a linear relationship between concentration and peak area in the range of 5-150 μg/mL with correlation coefficient > 0.999. Lamivudine was subjected to forced degradation studies under two conditions: mild and extensive stress testing. These studies included the effect of hydrolysis (neutral, acidic, and alkaline), oxidation, photolysis, and dry heat. The proposed method proved to be stability-indicating by the resolution of the drug from its forced degradation products, making use of DAD as a tool for peak identity and purity confirmation. By virtue of its sensitivity, the developed method was also extended to analyze lamivudine in biological fluids such as human plasma and urine with good recovery values (Graphical abstract).
Keywords
Lamivudine, Mild and Extensive Stress Testing, HPLC-DAD, Tablets, Plasma, Urine
Introduction
Lamivudine, 4-Amino-1-[(2R, 5S)-2-(hydroxymethyl)-1, 3-oxathiolan-5-yl] pyrimidin-2(1H)-one, is a nucleoside reverse transcriptase inhibitor structurally related to cytosine. It is converted intracellularly to the triphosphate, which halts the DNA synthesis of retroviruses, including HIV through competitive inhibition of reverse transcriptase and incorporation into viral DNA. It is also active against hepatitis B virus [1].
Lamivudine (LVD) is official in BP 2012 [2] and USP 38 [3] which describe HPLC methods for its determination as bulk powder. The quantification of Lamivudine in its pharmaceutical preparations and/or biological samples was described in numerous reports. Analytical methodology involved the use of techniques such as HPLC-UV using different types of columns [4-9], spectrophotometry either by direct absorbance measurement [10] or after reacting with different reagents such as ninhydrin, ascorbic acid and PBQ [11], iodate, methyl orange and indigo carmine [12]. Other analytical techniques were also adopted such as cyclic voltammetry, differential pulse voltammetry and square wave voltammetry [13].
Concerning stability-indicating assays and forced degradation studies, Kaul, et al. [14] reported the stress testing of lamivudine by subjecting it to acid and alkaline hydrolysis, oxidation, dry heat, wet heat and photo-degradation followed by HPTLC assay. Bedse, et al. [15] reported the forced decomposition behavior of lamivudine using LC-MS. Lamivudine has also been simultaneously determined in combination with other drugs either in biological matrices or in pharmaceutical preparations. In this context, lamivudine has been analyzed in binary mixtures with zidovudine by separation techniques such as liquid chromatography- tandem-mass spectrometry [16] solid phase extraction coupled to liquid chromatography/tandem-mass spectrometry [17] HPLC-UV [18], ion-pair HPLC [19], micellar electrokinetic chromatography [20] and HPTLC [21] and by other techniques such as derivative-differential UV spectrophotometry and compensation technique [22], in addition to first derivative spectrophotometry and first derivative of the ratio spectra [18].
In combination with stavudine, lamivudine has been determined by chemometric-assisted spectrophotometry [23]. Lamivudine has also been simultaneously determined with several other HIV nucleoside analogue reverse transcriptase inhibitors such as didanosine, abacavir, nevirapine and stavudine by RP-HPLC [24], capillary zone electrophoresis [25], HPTLC [26], and UV spectroscopy [26].
Our objective in the present work, was to develop a stability-indicating chromatographic method able to quantify the drug in dosage forms, plasma and urine (70% of the oral dose is excreted unchanged in urine) without interference from endogenous components or degradation products that are likely to be formed under the normal storage conditions (mild stress testing) and severe conditions (extensive stress testing).
Experimental
Instrumentation
The HPLC-DAD system consisted of Agilent 1200 series (Agilent Technologies, Santa Clara, CA, USA) (quaternary pump, vacuum degasser, and diode array and multiple, wavelength detector G1315 C/D and G1365 C/D) connected to a computer loaded with Agilent Chem Station Software. A Rheodyne manual injector with 20 μL loop was used.
Materials
An authentic sample of lamivudine was provided by European Egyptian Pharmaceutical Industries Co. and was used without any further purification. All chemicals and solvents were of HPLC grade. Phosphate buffer pH 4 was prepared according to USP 34 [3] using analytical grade of potassium dihydrogen phosphate and pH was adjusted using phosphoric acid. Urine and Plasma samples were donated from healthy volunteers.
General procedure
Preparation of stock standard solution of Lamivudine: The stock solution was prepared by dissolving lamivudine in purified water to obtain a final concentration of 5 mg/mL.
Development of the HPLC method and construction of calibration curve: The chromatographic separation was done using a C18 (octylsilyl) BDS Hypersil®column (4.6 × 150 mm × 5μm). The mobile phase was prepared by mixing acetonitrile and 50 mM phosphate buffer of pH 4 (adjusted using phosphoric acid) in a ratio of (10:90) v/v. The mobile phase was filtered and degassed by passing through a 0.45μm pore size membrane filter before use. The injection volume was 20 μL, the flow rate was 1.0 mL/min. and the U.V. detector was set at 280 nm. All measurements were performed at ambient temperature.
The working solutions were prepared by dilution of the standard solution of the drug with the mobile phase to reach concentration ranges of 5-150 μg/mL. Triplicate injections were made for each concentration and chromatographed under the optimized HPLC conditions. The calibration curve was constructed by plotting the peak areas against the corresponding concentrations.
Stability studies
Mild stress testing: Aliquots of 5 mL from LVD stock solution were transferred to 25-mL volumetric, flasks followed by 10 mL of purified water, and then drops of each of the following solutions were separately added:
1) 1 M HCl till pH 2
2) 1 M NaOH till pH 7
3) 1 M NaOH till pH 11
4) 1 M NaOH till pH 7, followed by 0.5 mL of 30% v/v hydrogen peroxide solution
The volume was adjusted in each flask to 25 mL with purified water. The solutions (1-3) were kept at room temperature in the dark for 14 days, while solution (4) was kept at room temperature in the dark for 24 hours. After the specified time, solutions (1) and (3) were neutralized to pH 7.0, then 2 mL aliquot from each solution was diluted to 20 mL with the mobile phase, filtered before injection into the HPLC column. The chromatograms were recorded and compared to a standard solution of the drug (of equal concentration) that has not been subjected to the previous treatment.
Extensive stress testing
Stress degradation by hydrolysis under acidic and alkaline conditions: Aliquots of 5 mL from LVD stock solution were transferred to 25-mL volumetric flasks and treated with 7.5 mL of 1M HCl or 1M NaOH, then heated in a boiling water bath at 100 ℃ for 2 hours. After the specified time, all solutions were neutralized to pH 7.0 and the volumes were adjusted to 25 mL using purified water. A 2 mL aliquot from each solution was diluted to 20 mL with the mobile phase and filtered before injection into the column. This procedure was performed in the dark in order to exclude the possible degradation caused by light exposure.
Oxidative degradation: Oxidative degradation was performed by heating 5 mL from LVD stock solution with 2 mL of 30% v/v of hydrogen peroxide in a water bath at 80 ℃ for 30 minutes. After the specified time, the volumes were adjusted to 25 mL using purified water. A 2 mL aliquot from each solution was diluted to 20 mL with the mobile phase and filtered before injection into the column. Oxidative degradation was also performed away from light.
Dry heat degradation: An amount of the drug powder (2 g) was kept in an oven at 90 ℃ for 8 hours. After the specified time 100 mg of the powder were accurately weighed and dissolved in 20 mL purified water. A 5 mL aliquot from this solution was diluted to 25 mL with purified water, and then a 2 mL aliquot was diluted to 20 mL with the mobile phase. The solution was filtered using 0.45 μm filter before injection into the column.
Wet heat degradation: Wet heat degradation was performed by heating 5 mL of LVD stock solution in a boiling water bath for 2 hours in the dark. After the specified time, the volume was adjusted to 25 mL using purified water. A 2 mL aliquot from this solution was diluted to 20 mL with the mobile phase and filtered prior to injection into the column.
Photolytic degradation: The Photo degradation study was performed by subjecting the stock solution of the drug to direct UV radiation (UV lamp set at 254 nm) at room temperature for 8 hours. After the specified time, 5 mL aliquot from the solution was diluted to 25 mL with purified water, and then a 2 mL aliquot was diluted to 20 mL with the mobile phase and filtered before injection into the column.
Application to pharmaceutical preparation
Lamidine® tablets (150 mg lamivudine/tablet): Ten tablets were weighed and finely powdered. An accurately weighed amount of the powder equivalent to 300 mg lamivudine was transferred to a 100-mL volumetric flask, extracted in 30 mL purified water, sonicated for 20 minutes then the volume was completed to 100 mL with purified water. The solution was filtered. Aliquots of the filtrate were diluted with the mobile phase to suit the concentration range of the method, filtered through 0.45 mm membrane prior to injection.
Application to spiked human plasma and urine: The stock solution of LVD was 100 folds diluted with purified water. Different aliquots from the diluted stock solution (to suit the concentrations in Table 1), were transferred into centrifugal tubes containing 4 mL plasma or urine. The tubes were shaken for 2 min. using a vortex tube shaker. Two mL acetonitrile were added, and the solutions were centrifuged for 30 min. at 8000 r.p.m. The centrifugates were transferred to clean centrifuge tubes and the residues were washed twice each with 1 mL of acetonitrile. The washing liquids were added to the centrifugates, which were diluted to 25 mL with the mobile phase and the procedure was completed as described under "general procedure".
Results and Discussion
Optimization of chromatographic conditions
Several parameters were optimized in order to achieve sufficient resolution between the intact drug peak and degradation products peaks with acceptable peak symmetry and retention times.
Mobile phase composition: Several mobile phases were tried using various proportions of different kinds of buffers and organic modifiers. Mobile phases composed of acetate buffer/methanol and acetate buffer/acetonitrile were tried but distorted peaks were obtained. A good symmetric sharp peak was obtained by using phosphate buffer/acetonitrile in a ratio of 90:10 v/v. By trying other ratios, less satisfactory results were obtained eg: At a ratio of (95:5) v/v the retention time was approximately doubled and at a ratio of 60:40 a bad distorted peak was observed. Ratios of 90:10 till 65:35 were found satisfactory.
Buffer concentration and pH: Phosphate buffers of different pH values ranging from pH 2 to 8 in 0.5-unit increment were investigated where a forked broad peak was obtained at pH 2 and 2.5.
A tailed peak was observed at pH 6, 6.5 and 7. Within the pH range of 3 to 5 the retention time remained constant, and a sharp peak of sufficient symmetry was obtained, therefore pH 4 was selected.
A buffer composed of 100 mg metaphosphoric acid and 200 mg potassium dihydrogen phosphate/liter was chosen for analysis. Changing the buffer concentration from half these values (50, 100 mg) to 10 folds (1000, 2000 mg) had no significant effect on retention times or peak shape or symmetry.
Flow rate: Different flow rates were studied 0.5, 0.8, 1.0, 1.5, 2, 2.5 mL/min. (All of which kept the column pressure within acceptable values). A Flow rate of 1.0 mL/min was selected for lamivudine, by virtue of the reasonable retention time achieved and its capacity to resolve the peak of the intact drug from those of the degradation products.
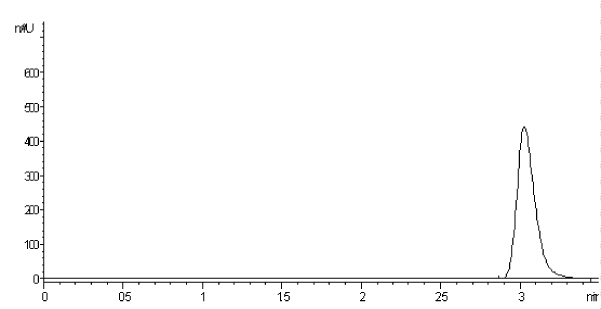
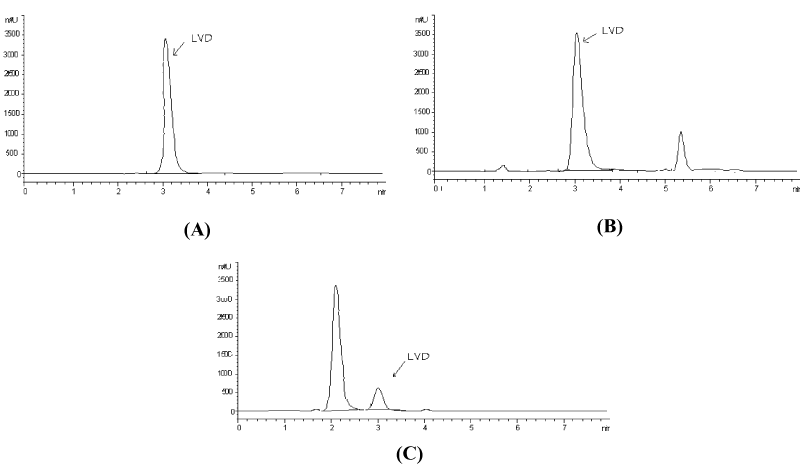
Using the above-mentioned optimized conditions, quantification was achieved using diode array detection based on peak area measurement. Figure 1 shows a typical chromatogram for LVD eluted at retention time 3.08 ± 0.059. Column performance (apparent efficiency) can be expressed by the number of theoretical plates (N) which equals about 1511.
Stability-indicating aspects
Forced degradation studies can help assess the stability-indicating power of the proposed analytical method. In this study, hydrolytic (using either HCl or NaOH solutions) and oxidative degradation (using H2O2) were performed at both ambient temperatures to simulate the normal shelf conditions (mild stress testing), and by heating (extensive stress testing).
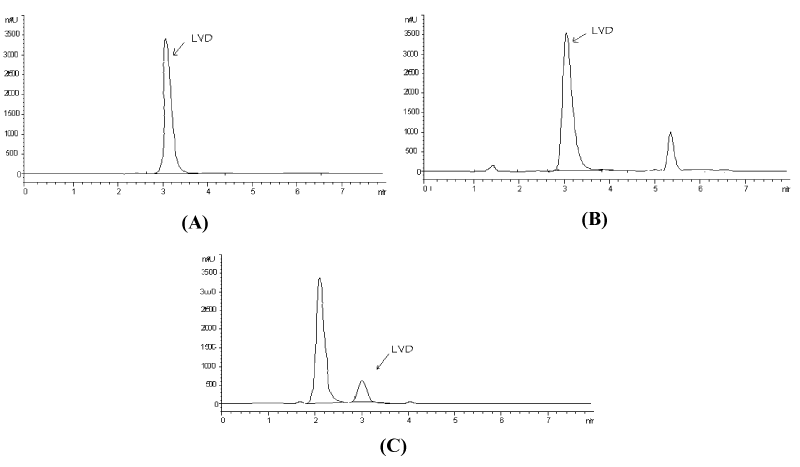
Effect of mild stress testing on lamivudine: No signs of degradation were observed in acid or neutral conditions at ambient temperature, as the analyte peaks showed peak areas identical to those of the standard peaks of the same concentrations. On the other hand, there was around 8.5% and 86% reduction in the peak area of lamivudine exposed to alkaline and oxidative degradation under mild conditions, respectively with the appearance of additional small peaks attributed to degradation products eluted at about 1.4 and 5.4 min for alkaline degradation and 2.1 min for oxidative degradation (Figure 2).
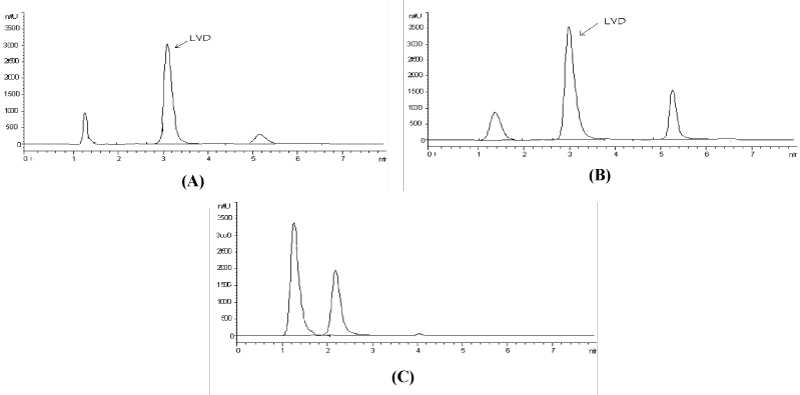
Effect of extensive stress testing on lamivudine: Lamivudine was subjected to forced degradation by acids, alkalis, heat, light, and oxidation, as per the ICH Q1A guideline. Lamivudine showed 18% degradation under acidic conditions, 32% under alkaline conditions and 100% under oxidative conditions (Figure 3), with the appearance of additional peaks attributed to degradation products eluted at about 1.4 and 5.4 min for acidic and alkaline degradations and at 1.2 and 2.2 min for oxidative degradation.
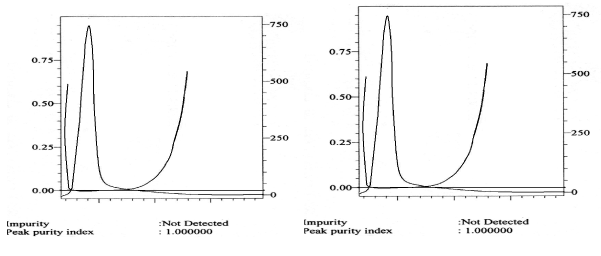
No degradation was observed after exposure to dry heat, wet heat, or UV light. Peak purity tests obtained from PDA confirmed that lamivudine peaks are homogenous and pure in all the samples subjected to stress conditions, thus confirming the stability-indicating power of the proposed method.
Validation of the proposed method
Linearity and concentration range: The linearity of the proposed HPLC method was assessed by analyzing different concentrations of the drug and generating the linear regression equation was by least square treatment of the calibration data. Under the optimized conditions, the measured peak areas were found to be proportional to the concentrations. Table 2 shows the linear regression equation, concentration range, correlation coefficient, variance of the intercept (Sa2), variance around the slope (Sb2). The value of the correlation coefficient indicates a good linearity.
Accuracy: The accuracy of the proposed method was evaluated by calculating the recoveries of the drug added to regular tablet excipients (lactose, magnesium stearate, microcrystalline cellulose, croscarmellose sodium and crospovidone), where good results were obtained (Table 2). The accuracy was further confirmed by comparing the results of the assay of the pharmaceutical preparation with the official B.P. HPLC method [2] by the student t- test (Table 3), where the calculated value was found smaller than the theoretical one.
Precision: The repeatability of the proposed method was appraised by calculating the relative standard deviation of the assay results of three different concentrations, each in three replicates. The results are shown in Table 2.
Selectivity: The selectivity of the proposed HPLC method was tested by applying the chromatographic method to the following solutions:
1. A solution of the tablet excipients (placebo). The chromatogram of such a solution showed no peaks at the retention time of the drug throughout the run time.
2. A Solution containing reference standard of LVD added to the solution of the tablet excipients (placebo). The chromatogram of such a solution was superimposable with that of the reference standard. The peak obtained was sharp and had a clear baseline separation.
An additional proof for the selectivity of the proposed HPLC method was obtained by testing the spectral purity of the eluted peak. The results indicated that the eluted peak was pure based on the following criteria:
∎ The spectra (A vs. λ) captured at different time intervals across the chromatographic peak of the drug with the photodiode array detector were overlaid for both the test and the reference solutions.
∎ The peak purity index was found to have a value very close to 1.
Limit of detection and limit of quantitation: The LOD is defined as the concentration that has a signal-to-noise ratio (S/N) of 3:1, while for the LOQ, the required ratio of S/N is 10:1. The calculated LOD and LOQ values for LVD by the proposed method are displayed in Table 2.
Robustness: Robustness was evaluated by making small variations in different conditions such as buffer pH (± 0.5 unit), detection wavelengths (±3 nm) and flow rates (± 0.1 mL/ min). These variations had no effect on the measured responses.
Analysis of pharmaceutical preparations
The developed HPLC method was utilized for the assay of LVD tablets. Sample preparation involved only a simple procedure with no need for the separation of the active pharmaceutical ingredient from the inactive additives. The diode array detector enables peak purity verification where no sign of co-elution from any of the inactive components was detected. The calculated recoveries revealed satisfactory accuracy and precision as indicated from % recovery, SD and RSD% (Table 3). The assay results of the developed method were compared to those of the B.P method [2] by the student t- test and the variance ratio, F test, where no significant difference was found between the two compared methods of analysis.
Analysis of lamivudine in plasma and urine
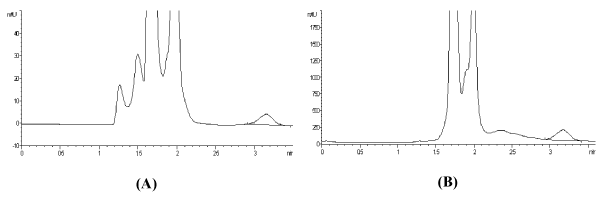
The developed method was extended to the analysis of lamivudine in biological fluids such as plasma and urine. Acceptable results were obtained in both matrices as evidenced by the % recoveries that ranged from 89 to 95% (Table 1). Additionally, the chromatograms showed no interference in the form of co-elution at the retention time of the drug (Figure 4A and Figure 4B, and this was confirmed by recording the corresponding purity plots (Figure 5A and Figure 5B.
Conclusion
An isocratic HPLC method has been developed for the determination of Lamivudine in bulk, dosage forms, plasma, and urine. The advantages of the proposed method over the previously reported ones are the use of a DAD detector that is widely available in the ordinary laboratories, with no need for the more sophisticated mass detector. Another noted advantage is its stability-indicating power and its ability to selectively analyze the studied drug in the presence of its forced degradation products generated under mild or extensive stress conditions, and in the presence of endogenous plasma and urine components.
Conflict of Interest and Funding
The authors declare no conflict of interest and no fund was received.
References
- Sweetman SC (2009) Martindale, The Complete Drug Reference,36th edition, The Pharmaceutical Press, USA.
- The British pharmacopoeia (1980) Her Majesty's Stationery Office, London, 1243, 1370-1371, 1837-1838, 2080-2081.
- Rockville MD (2015) The United States Pharmacopeia (38th edn), The National Formulary, (33th edn) The Official Compendia of Standards, United States Pharmacopeial Convention, Inc., Asian Edition.
- Shao LH, Ma LX, Sun SA, Pan Y Z, Wang XT (2002) Yaowu Fenxi Zazhi 22: 53-55.
- Aboul-Enein HY, Hefnawy MM (2003) High throughput analysis of lamivudine in pharmaceutical preparations using monolithic silica HPLC column. Anal Lett 36: 2527-2538.
- Ozkan SA, Uslu B (2002) Rapid HPLC assay for lamivudine in pharmaceuticals and human serum. J Liq Chromatogr Related Technol 25: 1447-1456.
- Patro SK, Swain SR, Patro VJ, et al. (2010) Development and validation of high performance liquid chromatographic method for determination of lamivudine from pharmaceutical preparation. E-Journal of Chemistry 7: 117-122.
- Patil VD, Chaudhari TR, Pendse US, et al. (2008) RP-HPLC method for determination of lamivudine in tablet. ACAIJ 7: 240-244.
- Sarkar M, Khandavilli S, Panchagnula RJ (2005) Development and validation of RP-HPLC and ultraviolet spectrophotometric methods of analysis for the quantitative estimation of antiretroviral drugs in pharmaceutical dosage forms. Chromatogr B Anal Technol Biomed Life Sci 830: 349-354.
- Pimpodkar NV, Mahajan NS, Jadhav RL, et al. (2008) New Spectrophotometric Method for Estimation of Lamivudine in Bulk and Pharmaceutical Dosage Forms. Int J Chem Sci 6: 688-692.
- Raju NA, Begum S, Balamami S, et al. (2008) Acta Ciencia Indica Chemistry 34: 489-492.
- Basavaiah K, Somashekar B C, Ramakrishna V (2007) Rapid titrimetric and spectrophotometric assay methods for the determination of lamivudine in pharmaceuticals using iodate and two dyes. Anal Chem 62: 542-548.
- Dogan B, Uslu B, Suzen S, et al. (2005) Electrochemical evaluation of nucleoside analogue lamivudine in pharmaceutical dosage forms and human serum. Electroanalysis 17: 1886-1894.
- Kaul N, Agrawal H, Paradkar AR, et al. (2004) The International Conference on Harmonisation Guidance in Practice: Stress degradation studies on lamivudine and development of a validated specific stability-indicating HPTLC assay method. Chromatographia 60: 213-221.
- Bedse G, Kumar V, Singh S (2009) Study of forced decomposition behavior of lamivudine using LC, LC-MS/TOF and MSn. J Pharm Biomed Anal 49: 55-63.
- Kenney KB, Wring SA, Carr RM, et al. (2000) Simultaneous determination of zidovudine and lamivudine in human serum using HPLC with tandem mass spectrometry. J Pharm Biomed Anal 22: 967-983.
- De-Cassia-EEstrela R, Salvadora MC, Suarez-Kurtz G (2004) Rapid Comm Mass Spectrom 18: 1147-1155.
- Uslu B, Ozkan A (2002) Determination of lamivudine and zidovudine in binary mixtures using first derivative spectrophotometric, first derivative of the ratio-spectra and high-performance liquid chromatography-UV methods. Anal Chim Acta 466: 175-185.
- Fan B, Stewart JT (2002) Determination of zidovudine/lamivudine/nevirapine in human plasma using ion-pair HPLC. J Pharm Biomed Anal 28: 903-908.
- Sekar R, Azhaguvel S (2005) Simultaneous determination of HIV-protease inhibitors lamivudine and zidovudine in pharmaceutical formulations by micellar electrokinetic chromatography. J Pharm Biomed Anal 39: 653-660.
- Habte G, Hymete A, Mohamed AI (2009) Simultaneous separation and determination of lamivudine and zidovudine in pharmaceutical formulations using the HPTLC method. Anal Lett 42: 1552-1570.
- Erk N (2004) Derivative-differential UV spectrophotometry and compensation technique for the simultaneous determination of zidovudine and lamivudine in human serum. Pharmazie 59: 106-111.
- Abd El-Maaboud IM, Mikre W (2009) Determination of lamivudine and stavudine in pharmaceutical preparations using chemometrics-assisted spectrophotometry. Saudi Pharm J 17: 275-281.
- Rezk NL, Tidwell RR, Kashuba ADM (2003) Simultaneous determination of six HIV nucleoside analogue reverse transcriptase inhibitors and nevirapine by liquid chromatography with ultraviolet absorbance detection. J Chromatogr. B Anal Technol Biomed Life Sci 791: 137-147.
- Fan B, Stewart JT (2002) Determination of lamivudine/didanosine/nevirapine in human serum using capillary zone electrophoresis. Journal of Capillary Electrophoresis 7: 103-106.
- Anbazhagan S, Indumathy N, Shanumugapandiyan P, et al. (2005) Simultaneous quantification of stavudine, lamivudine and nevirapine by UV spectroscopy, reverse phase HPLC and HPTLC in tablets J Pharm Biomed Anal 39: 801-804.
Corresponding Author
Rim S Haggag, Department of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, University of Alexandria, El-Messalah, Alexandria 21521, Egypt, Tel: +20-3-487-1317, Fax: +20-3-487-3273.
Copyright
© 2021 Haggag RS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.