N-Acetylcysteine: A Natural Antidote for Alzheimer's Disease
Abstract
Alzheimer's disease (AD) is the most devastating age-related dementia, which has no effective treatment. Since the pathological hallmarks of AD brains are Aβ plaques and intra-neuronal tau-containing neurofibrillary tangles, most therapeutic approaches, including immunotherapy, are based on the concept that the accumulation of these proteins produces neuronal damage and death. Alternatively, it has been proposed that reactive species can cause oxidative damage including mitochondrial disruption, contributing to AD initiation and progression. Can these cellular dysfunctions be linked by a common pathogenic mechanism susceptible to therapy with the natural compound N-acetylcysteine? A cellular cysteine network (CYSTEINET) has been proposed as a functional and structural matrix of interconnected sensitive cysteine-containing proteins (SCCPs) that in conjunction with reactive species and the cysteine/glutathione cycles can regulate the bioenergetic metabolism, the redox homeostasis, and the cellular survival through different pathways that bear the same regulatory thiol radical. The present paper proposes that cysteinet is impaired in Alzheimer's disease resulting in a functional and structural deregulation of the matrix of interconnected cysteine-containing proteins that result in misfolding, aggregation and accumulation of specific toxic proteins. In this context, the role of N-acetylcysteine to prevent and restore cysteinet deregulation in AD development and progression is discussed.
Keywords
Cysteine, Thiol, Neurodegeneration, Reactive species, Redox homeostasis, Acetylcysteine, Alzheimer, Cysteinet
Introduction
Alzheimer's disease (AD) is the most devastating neurodegenerative disorder accounting for 60% to 70% cases of dementia. Most AD cases are sporadic and occur in elderly people over 60 years old. Therefore, like in Parkinson's disease (PD), the principal risk factor for sporadic AD is aging. Cognitive impairments of patients with AD are associated with synaptic deficits and neuronal loss, which have been shown early events in the disease [1-3]. The prevalent hypothesis explaining the physiopathology of AD is the amyloid cascade, which postulates that the aberrant processing of amyloid precursor protein (APP) through amyloidogenic [3-6] pathways yield increased Aβ1-42 peptide that is the responsible for neurotoxicity, neuronal loss and cognitive impairment in AD patients [7,8]. In addition, intracellular neurofibrillary tangles are formed through the hyperphosphorylation of the microtubule-associated protein tau, which forms part of the cytoskeleton of neurons [9,10].
Since the pathological hallmarks of AD brains are Aβ plaques and intracellular tau-containing neurofibrillary tangles, most therapeutic approaches have been directed towards the inhibition of the aggregation and accumulation of these proteins in the brain. The food and drug administration (FDA) has approved various drugs for AD treatment that can be divided into compounds that directly target amyloid Aβ by active and passive immunization, those targeting the inhibition or modulation of the APP γ-secretase enzyme, and those targeting the APP β-secretase enzyme (BACE1 inhibitors drugs), which have shown controversial and no conclusive results [11,12].
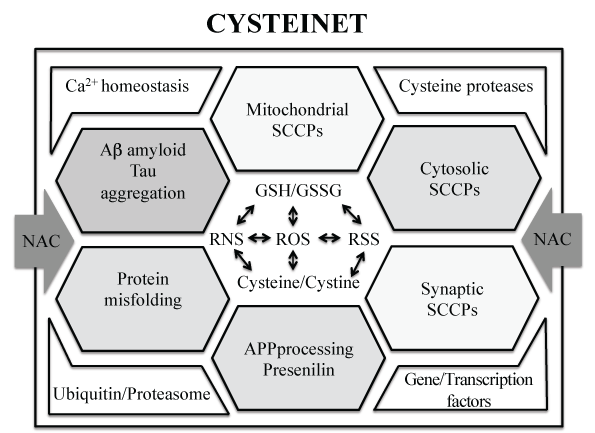
In view of the above, other theories have been elaborated to explain AD etiology and physiopathology including various attempts to integrate the many pieces that form part of the AD puzzle. Our approach is that AD physiopathology is better explained if diverse etiologic factors (genetics, mitochondrial, toxic, metabolic, inflammatory, and age-related) isolated or in conjunction, initiate some biochemical disturbances that converge in a common pathway ending in neurodegeneration. Cysteinet has been proposed as a hierarchical bottom-up biochemical network, composed of
(a) Reactive species acting as molecular massagers and modulators,
(b) Cysteine/cystine and reduced/oxidized glutathione (GSH/GSSG) cycles, and
(c) All peptides and proteins that have "sensitive" cysteine residues in their structure [13,14].
This interconnected matrix of sensitive cysteine-containing proteins (SCCPs) has very different metabolic, signaling and structural functions in the cell but is regulated by the same thiol radical. These SCCPs use cysteine residues as cellular sensors, which can entangle, at very short timescale, different cellular functions. The central concept is "sensitive cysteines", which are all cysteine residues in peptides and proteins that can suffer redox modifications (s-glutathionylation, s-nitrosylation, sulfenylation, disulfide bonds formation) under physiological or pathological conditions resulting in a reversible or irreversible change in the protein function [15,16]. Therefore, it is proposed that cysteinet deregulation is the common mechanism through which different etiologic events or insults overcome age-associated cellular redox homeostasis and evolve toward a vicious circle in age-related neurodegenerative diseases. For example, the ROS overproduction associated with age-related mitochondrial DNA damage and the subsequent impairment in the bioenergetic ability in patients that are homozygote for apolipoprotein E4 may result in a dysfunction of cysteinet regulation that can entangle diverse pathways facilitating, among others, Aβ amyloid and tau aggregation (Figure 1). In this regard, we propose that cysteinet deregulation in AD can be prevented and restored by the chronic administration of NAC counteracting the deleterious redox modification of many SCCPs in different pathways.
In the next sections, it has been reviewed the critical role played by redox modification of thiol groups in SCCPs from diverse cellular pathways, and how they can be integrated into the proposed cysteinet deregulation that may explain AD physiopathology.
Risk Factors and Cysteinet Deregulation in AD
Aging
Aging is the major risk factor for the development of late-onset AD. Aging is closely associated with oxidative damage to cellular macromolecules produced by reactive species [17-19]. A study in healthy humans aged 19-85 have shown an age-related linear decrease in the cysteine/cystine ratio throughout life, and an age-related decrease in the total glutathione content and GSH/GSSG ratio after the age of 45 [20]. This show an age-related shift of redox homeostasis toward pro-oxidant conditions, which have critical consequences, not only in many protective antioxidant enzymes that use GSH to counteract the deleterious effects associated with aging [21], but also in many regulatory pathways that depend on the functional integrity of SCCPs [22,23] including mitochondrial ones [24,25]. Therefore, aging increases the rate of irreversible damage to structural and metabolic proteins interfering with normal cellular functions and homeostasis. New quantitative proteomic methods indicate that aging is associated with a shift in redox-regulated proteins resulting in major changes in the glycolytic enzymes as well as in the regulatory enzymes controlling the energy homeostasis in muscular post-mitotic cells [26]. Besides, recent investigations have shown that normal brain aging is characterized by a deregulated pattern of expression of specific aggregation-prone proteins that predispose to Aβ and tau deposition, in contrast to the suboptimal expression of homeostatic proteins [27]. These results suggest that there is an increased susceptibility to protein aggregation in specific brain areas more vulnerable to AD development [27].
In agreement with previous data [28,29], genome-wide gene expression studies have provided evidence of reduced mitochondrial function during aging [30,31] and reduced expression of genes involved in mitochondrial energy metabolism in humans with cognitive decline and AD [32].
Finally, it seems that caloric restriction decreases the body temperature, the rate of metabolism, and the production of reactive species counteracting age-related oxidative damage [33]. Likewise, caloric restriction can stabilize mitochondrial function reducing oxidative damage in neurons in response to different types of genetic and environmental factors [34]. Therefore, age-associated oxidative modification of SCCPs may represent the Achilles' heels for senescence and age-associated neurodegenerative disorders [35,36].
Apolipoprotein E
The most well known genetic link to late-onset AD is the apolipoprotein (APO) genotype E4 (APOE4) [37]. Individuals with an E4 allele have a reduced lifespan [38] and they have an increased risk to develop AD [39]. There are three alleles of apolipoprotein E that differ in two amino acids. APOE2 contains a cysteine in each position, APOE3 contains a cysteine in one of the positions, but APOE4 does not contain a cysteine in either position. The mechanism by which APOE4 accelerates brain aging may involve decreased antioxidant and neuroprotective properties of this apolipoprotein isoform, since cysteine residues in APOE2 and APOE3, may bind to and detoxify 4-hydroxynonenal, a cytotoxic product of lipid peroxidation [40]. Therefore, apolipoprotein E isoforms have different potential sites for redox modification and regulation through sensitive cysteine residues. Indeed, apolipoprotein E isoforms can bind to neuronal nitric oxide synthase (NOS) and they can be S-nitrosylated in human hippocampus, possibly regulating the lipid metabolism in AD [41].
Cysteinet Deregulation in AD
Post-translational oxidative modifications of proteins have diverse functional implications including the control of the redox microenvironment and homeostasis. Since the thiol group of cysteine has a number of oxidation states, it is highly sensitive to environmental, biochemical, and pathological conditions resulting in the reversible or irreversible oxidation of this radical. Therefore, redox modifications of SCCPs can stabilize proteins against further irreversible oxidative damage, or they can be reverted to its physiological state [42]. The reversibility of thiol modifications allows that cysteine residues can function as switches in many regulatory pathways (Figure 1) as well as at many structural sites into the cell (Table 1). However, the irreversibility of thiol oxidation produces cumulative damage to proteins with diverse functional consequences [43,44].
Reactive species, mitochondrial SCCPs and cysteinet deregulation in AD
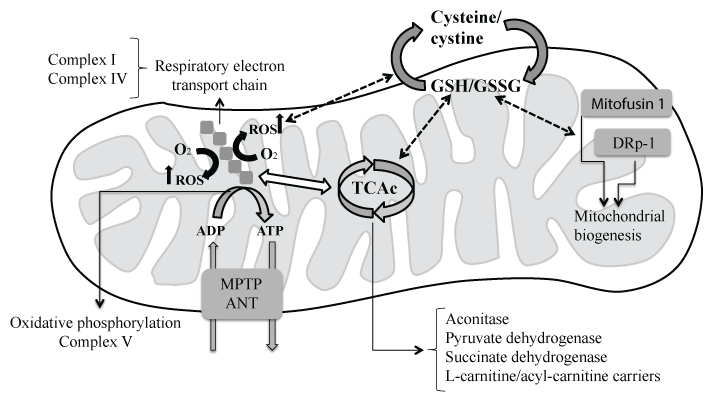
Physiological concentrations of reactive species (reactive oxygen species (ROS), reactive nitrogen species (RNS) and reactive sulfur species (RSS)) contribute to regulating cell metabolism and signaling pathways [45,46]. However, AD brains exhibit increased levels of reactive species, which affect proteins that are critical to neuronal survival contributing to the disease physiopathology [47]. In this regard, mitochondrial dysfunction plays a key role in AD physiopathology [48], occurring prior to the onset of clinical symptoms and Aβ plaque formation [48-52]. Indeed, synaptic mitochondria seem particularly vulnerable in aging and AD [52-54]. Mitochondria are the principal source of reactive species, which can modulate many SCCPs into the organelle that participate in the bio-energetic capacity including enzymes of the tricarboxylic acid cycle (TCAc) and enzymatic complexes of the respiratory electron transport chain and oxidative phosphorylation (Figure 2).
Aconitase and pyruvate dehydrogenase can be reversibly inactivated by hydrogen peroxide or S-glutathionylation through the oxidation of sensitive cysteine residues [15,42]. Succinate dehydrogenase can be modified by S-glutathionylation, and L-carnitine/acyl-carnitine carriers have also been found to be S-glutathionylated on Cys136 and Cys155 [15]. The oxidative modification of these enzymes by S-glutathionylation is reversible, and it occurs under physiological conditions. However, during the aging process, when GSH/GSSG ratio into de mitochondria is low, the oxidative modification of these SCCPs may be irreversible (Figure 1 and Figure 2).
Complex I of the mitochondrial respiratory chain can undergo sulfenylation, which decreases its activity. Sulfenylation may suffer further oxidation that irreversibly deactivates Complex I [15]. However, it seems that cysteine residues that are oxidized in Complex I can be protected from further oxidation by S-glutathionylation [42]. This S-glutathionylation of Complex I limits NADH production, and it decreases electron flow through the respiratory chain diminishing ROS generation, which protects this enzymatic complex from irreversible oxidation by reactive species (Figure 2). However, recent data have shown a disruption of Complex I activity at late stages of dysfunction in a mouse model of AD [55].
Complex IV (cytochrome c oxidase) is a key enzyme in the respiratory electron transport chain of the mitochondria, and it has critical cysteine residues that play fundamental roles in metal coordination necessary for the redox-linked proton pumping by the enzyme [56,57]. There is extensive evidence for reduced complex IV activity in AD brain tissue, fibroblasts and blood platelets [58-61].
Oxidative phosphorylation is also modulated by redox modification of the ATP synthase (Complex V) that can be S-glutathionylated on Cys294 of the α-subunit located in the F1 hydrophilic part of the Complex. Cys294 may also react with the neighboring Cys103 residue to form a disulfide bridge [62]. S-glutathionylation blocks the nucleotide binding to the complex, which results in a decrease in the production of ATP. Therefore, the oxidative modifications of SCCPs can control the mitochondrial ROS generation and the bioenergetic capacity of the cell (Figure 2).
Energy demand in neurons depends on mitochondrial dynamics mediated by fission and fusion mechanisms to generate new mitochondria. In this regard, recent studies have shown that β-amyloid accumulates inside AD brain mitochondria interfering with mitochondrial fusion and fission contributing to the development of AD [63,64]. Among SCCPs that participate in the mitochondrial biogenesis, the integral membrane GTPases Mitofusin 1 [65] and the dynamin family of GTPases dynamin-related protein-1 (Drp-1) are two well-studied examples. S-nitrosylated Drp1 levels are significantly increased in the brain of AD patient compared to controls, and it has been associated with the anomalous fragmentation of mitochondria and the consequent loss of synapses [66,67]. Besides, S-glutathionylation of mitofusin proteins is required to induce mitochondrial hyperfusion, which is regulated by redox micro-environmental conditions [68,69]. Therefore, mitochondrial SCCPs that regulate the structure and biogenesis of the organelle are dependent on the redox modifications of thiol groups in cysteine residues (Figure 2).
Finally, reactive species overproduction can modify the mitochondrial permeability transition pore (MPTP), which is a non-selective pore that allows the free diffusion of molecules under 1.5 KDa in size [70]. Sulfenylation and S-glutathionylation can induce programmed cell death in the mitochondria through the modulation of MPTP opening [70] that require the disulfide bond formation between specific cysteine residues in the adenine nucleotide translocator (ANT). ANT is a structural component of MPTP [70,71], which exports ATP from the mitochondrial matrix and imports ADP into the matrix [72].
Cytosolic SCCPs and cysteinet deregulation in AD
Many extra-mitochondrial proteins are regulated by cysteine redox modifications including many proteins involved in the glucose metabolism such as glyceraldehyde-3-phosphate dehydrogenase, pyruvate kinase, phosphofructokinase, glucose 6-phosphate isomerase, glycogen phosphorylase, phosphoglycerate mutase 1, and phosphoglucomutase 2, which require cysteine residues for their activity [73] (Figure 1 and Table 1).
A well-studied example of the glycolytic pathway that has been implicated in AD physiopathology is the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [74]. GAPDH catalyzes the reversible phosphorylation of G3P, which involves the thiol group of Cys152 [74]. Sensitive cysteine residues of GAPDH are oxidized by hydrogen peroxide, which decreases the stability of the protein, giving rise to monomers, dimers, and other denatured GAPDH products [74]. Furthermore, under pathological conditions glutathione can react with sensitive cysteine residues of GAPDH forming disulfide bonds [74]. GAPDH has been found oxidized in AD brain [74], although some studies have suggested that S-glutathionylation of GAPDH is a mechanism against permanent damage of the protein in the oxidizing environment of the AD brain [74]. Besides, GAPDH can be reversibly inhibited by s-nitrosylation of sensitive cysteine residues by nitric oxide, which can inhibit their dehydrogenase activity [74].
Protein tyrosine kinases (PTK) are a family of enzymes that contain a conserved sensitive cysteine residue. For example, protein tyrosine phosphatase 1b is redox regulated by cysteine residues placed in the catalytic center of the enzyme, which is reversibly oxidized by hydrogen peroxide to form sulfenic acid as well as by S-glutathionylation [75]. The result of these oxidative changes is an increase in tyrosine phosphorylation [75].
Synaptic proteins and cysteinet deregulation in AD
Cysteine string protein α (CSPα) is a protein involved in neurodegeneration [76] that regulates vesicle endocytosis, which participates in the regulation of synaptic transmission, and in the maintenance of synapses. CSPα contains a cysteine-rich "string" region for attachment to the synaptic vesicles that depend on its oligomerization [76]. Recently it has been reported that the expression of CSPα is reduced in degenerating areas of the forebrain in post-mortem samples of AD [77].
Synaptophysin is a membrane protein that forms channels in the synaptic vesicle, which depend on sensitive cysteine residues. The molecule contains unstable disulfide bonds that cross-linking with neighboring synaptophysin monomers. Cross-linking of synaptophysin by disulfide bonds are triggered by redox modification, suggesting that native synaptophysin depends on the adequate formation of cysteine associated disulfide bonds to form multimeric complexes in the adequate phospholipid environment [78]. This important protein plays a key role in the synaptic alterations associated with AD and its loss in the hippocampus has been correlated with cognitive decline in AD patients [79]. Besides, synaptophysin concentrations in the frontal cortex of AD patients were significantly lower than in controls [80].
More than 50% of AD patients exhibit α-synuclein accumulation [81]. Although human α-synuclein does not contain any cysteine residues [82], tyrosine to cysteine substitution at critical positions in the α-synuclein protein can increase dimer formation and accelerate protein aggregation and cellular toxicity of α-synuclein [83]. Through a chaperone-like function, α-synuclein may act in synergy with CSP-α in the assembly of the SNARE (soluble NSF attachment protein receptor) complex [84], and this effect involve the binding to synaptotagmin proteins, which have critical cysteine residues in their structure [84,85] (Figure 1).
APP processing, Aβ aggregation and cysteinet deregulation in AD
In general, there are two clinical forms of AD, the familial or early-onset AD (5% of all AD cases) produced by mutations in APP, presenilin-1 and presenilin-2 genes [86,87], and the highly prevalent sporadic or late-onset AD (95% of AD cases). The discovery that transgenic mice expressing familial human APP and presenilin mutations showed the major features of the human disease [88] contributed to establishing the amyloid cascade hypothesis [89]. Mutations in early-onset cases result in the aberrant processing of APP, leading to the aggregation of Aβ peptides and the subsequent hyperphosphorylation of tau, which seem to be critical factors for AD development [88].
Human presenilin-1 contains five native cysteine residues [90], three cysteine residues are in the transmembrane domains and two are exposed to the cytosol. Cys410 and Cys419 are in the presenilin-1 transmembrane domain 8, and both of these cysteine residues can crosslink with Cys92 in the transmembrane domain 1, contributing to conformational changes [91] that allow the interaction with the active site of γ-secretase, suggesting that these sensitive cysteine residues may support the integrity and hydrophilic environment of this active site [91]. This may have relevance in AD physiopathology since familial cases have shown mutations at three of the five endogenous cysteine residues in presenilin-1 [92,93], which would initiate cysteinet deregulation in early onset cases (Figure 1). On the other hand, disulfide bridges between residues Cys14-Cys31 and Cys56-Cys65 in presenilin-2, which are absent in the same region of presenilin-1, are critical determinants of the tertiary structure of presenilin-2. Interestedly, these sensitive cysteine residues seem to be important in the regulation of ryanodine receptor (RYR)-mediated Ca2+ release [94].
Moreover, protein palmitoylation consists of a thioester linkage between a palmitic acid and a cysteine residue to increase the hydrophobicity of the protein facilitating its integration in membranes [95]. APP has two palmitoylation sites at Cys186 and Cys187 that are required for its exit from the endoplasmic reticulum (ER). Cys186 and Cys187 reside within the copper-binding domain of APP, and they can stabilize the domain structure by forming disulfide bonds with Cys158 and Cys133, respectively [96]. Indeed, increased APP palmitoylation may enhance non-amyloidogenic β-cleavage of APP [96].
Therefore, under the view of the present hypothesis, mutations in APP, presenilin 1 or presenilin 2 are the etiologic factors that initiate cysteinet deregulation, which perpetuates and spreads the disequilibrium in the redox homeostasis in cases of early-onset AD (Figure 1).
Tau protein aggregation and cysteinet deregulation in AD
Twenty years ago it was demonstrated that pathological aggregation of tau protein into paired helical filaments (PHFs) and neurofibrillary tangles are dependent on the intermolecular cross-link of Cys322 [97]. The authors suggested that in addition to the disequilibrium between protein kinases and phosphatases, there is an age-related increase in neuronal oxidative stress that leads to the hyperphosphorylation of the protein (Figure 1). Indeed, it has been shown that peroxynitrite and hydrogen peroxide can induce cysteine oxidations in tau and microtubule associated protein-2 (MAP2) altering the ability of these proteins to assemble with microtubules [98]. Therefore, increased oxidative modification of sensitive cysteine found in brain aging [97] would provide a link for the aggregation of both types of AD deposits, since the amyloid deposits also appear to be related to increased oxidation [97].
Biochemical and kinetic studies indicate that tau catalyzes auto-acetylation mediated by a pair of catalytic sensitive cysteine residues in its microtubule-binding domain [99]. Tau cysteine residues are required for auto-acetylation increasing insoluble tau accumulation by both intra- and intermolecular acetylation mechanisms (Table 1). In fact, it is estimated that tau proteins are about 99% bound to microtubules in mature neurons [99], which inhibit tau acetyl-transferase activity by blocking the cysteine residues that are linked to microtubules. Therefore, it has been suggested that microtubule detachment and activation of tau auto-acetyl transferase activity could represent a pathological event, in which continual self-acetylation gradually shifts the tau-microtubule binding equilibrium toward cytosolic tau accumulation, providing an increased pool of aggregation-prone tau species [99]. Consistent with this model, the levels of acetyl-CoA are reported to be elevated in AD brain [99], which would support increased tau acetyl-transferase activity during the disease progression. Interestingly, methylthioninium, a tau aggregation inhibitor, can prevent the formation of tau filaments and their toxic precursors through oxidation of cysteine residues in the tau repeat domain preventing the formation of disulfide bridges and retaining the tau protein in a monomeric conformation [100]. Additionally, it has been demonstrated that compounds that bind cysteine residues of tau can prevent neurofibrillary tangle-associated brain dysfunction [101].
Calcium homeostasis and cysteinet deregulation in AD
Calcium (Ca2+) disturbances in AD have been attributed to Aβ amyloid up-regulation [102] trough the formation of channels in membranes, up-regulation of NMDARs channels, and a mechanism that implicates the prion protein [103]. However, intracellular Ca2+ levels in neurons depend on the equilibrium between pumping of cytosolic Ca2+ into the ER lumen by sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) pump, and the release of Ca2+ from the ER through the activation of two types of Ca2+ receptors, the inositol 1,4,5-trisphosphate receptor (IP3R) and the RYR (Figure 1). One function of RYR is to amplify the IP3R-mediated release of Ca2+ from ER enhancing neuronal Ca2+ signals [104]. IP3R and RYR receptors are the most expressed intracellular Ca2+ channels regulated by thiol groups of cysteine. RYR1 channel has multiple cysteine residues that can suffer S-nitrosylation and S-glutathionylation, which can destabilize the closed state of the RYR1 complex resulting in an altered release of Ca2+ to the cytoplasm [105]. Similarly, IP3R contain multiple sensitive cysteine residues that serve as the target of ROS produced in both the ER and the mitochondria [106]. This receptor regulates the cytoplasmic Ca2+ levels in cells through the modulation of specific cysteine residues within the IP3-binding core and suppressor domain of the protein, changing the active conformation of the receptor [107]. These findings may be relevant having into account that IP3R levels have been found decreased in the hippocampal formation of AD patients, showing significant correlations with senile plaque staging and neurofibrillary tangle pathology [108]. Besides, high glucose levels can oxidize SERCA Cys674 preventing its inhibition by nitric oxide and hydrogen peroxide [109], which can contribute to Ca2+ disturbance. Interestingly, quantitative mapping of oxidation-sensitive cysteine residues in SERCA from skeletal muscle of Fisher 344 x Brown Norway F1 hybrid rats, were found selectively oxidized during aging [110]. These data suggest an age-related loss of sensitive cysteine residues in SERCA explaining the age-associated decrease in the specific Ca2+-ATPase activity [110].
Protein misfolding and cysteinet deregulation in AD
Under physiological conditions, chaperones help proteins to achieve their three-dimensional (3-D) configuration, but aging and other stressing conditions can disturb the exquisite balance among the synthesis, folding and degradation of proteins resulting in their accumulation. Protein folding depends on the amino acid primary sequence as well as on the cysteine content to form a disulfide bond under different redox conditions [111] (Figure 1). Cysteine thiol groups can switch between oxidized and reduced states contributing to protein folding [112]. In fact, many misfolded proteins contain disulfide bonds [113]. The spontaneous folding process can be particularly slow in the case of cysteine-containing proteins that require the formation of disulfide bonds [114]. Therefore, in vivo disulfide bond formation is catalyzed by specialized enzymes such as protein disulfide isomerase (PDI) [115]. This ER enzyme has been found S-nitrosylated in brain samples of sporadic Parkinson and Alzheimer's patients [116]. S-nitrosylation of PDI facilitates further oxidative modifications compromising chaperone/protein folding and accumulation in ER [116].
Mitochondrial biogenesis depends on protein import from the cytosol through mitochondrial translocation pathways (Table 1). Among these pathways, the mitochondrial import and assembly (MIA) pathway targets proteins to the intermembrane space (IMS) and couples import to folding and oxidation resulting in the covalent modification of the incoming protein precursor that incorporates disulfide bonds in the process [117]. For example, Mia40 is a thiol oxidase does not belong to the thioredoxin family, and its catalytic disulfide is arranged in a unique Cys-Pro-Cys motif. The formation of the initially mixed disulfide between Mia40 and a substrate protein directs the course of its oxidative folding [118], which seem dependent on specific cysteine residues to form the initial enzyme-substrate-mixed disulfide [118]. Therefore, Mia40 and other redox active proteins like thioredoxin, glutaredoxin, and peroxiredoxin into the IMS have been suggested as redox-dependent chaperone-like mechanisms in mitochondria associated with sensitive cysteine residues [118] (Figure 1).
Ubiquitin-proteasome pathway and cysteinet deregulation in AD
Oxidative damage in proteins and aggregates are recognized and degraded by the ubiquitin-proteasome machinery in a first step to recycling those proteins [119]. The ubiquitin-proteasome machinery is a multistep pathway that involves the activation of the ubiquitin protein through an ATP-dependent process that uses two enzymes (E1 and E2) to form ubiquitin-E2, which binds to target protein by the mediation of the ubiquitin protein ligase (E3) [120]. These steps are dependent on cysteine residues that form consecutive thioester linkages ending in protein degradation in the proteasome [120] (Figure 1). Therefore, the ubiquitin-proteasome multistep pathway is dependent on the physiological redox regulation of thiol groups of sensitive cysteine residues of the proteins implicated in the pathway [121,122]. Accumulation of ubiquitinated protein aggregates occur during normal brain aging and reach pathological levels in neurodegenerative disorders such as AD [123]. A recent study in a mouse model of reduced proteasome activity in the brain showed increased concentrations of those proteins that are elevated in the brain of AD patients. Furthermore, these mice showed deficits in spatial memory suggesting that defective proteasome function participates in the cognitive impairment [124].
Cysteine proteases and cysteinet deregulation in AD
Emerging evidence shows that cysteine proteases play important roles in AD physiopathology including calpains, cathepsins, and caspases [125].
For example, calpains are cysteine proteases that have been implicated in AD through hyperactivation, which results from several factors, including enhanced intracellular Ca2+ concentration [126]. Besides, Aβ peptides induce calpain activation by elevation of Ca2+ levels leading to neuronal cell dysfunction and death [125]. Activation of the catalytic center of calpains 1 and 2 is dependent on Ca2+ allowing the rearrangement among the amino acids cysteine, histidine, and asparagine [127]. Therefore, calcium binding to the protein and the proper oxidation state of sensitive cysteine residues in the catalytic center are necessaries to the adequate activities of these proteases [128].
The role of caspases in AD physiopathology has been widely examined, and many proteins of the caspase family are transcriptionally elevated in AD [125]. In this regard, nitric oxide may nitrosylate sensitive cysteine residues on the catalytic center of caspase-3 preventing apoptosis in AD [129]. Besides, inhibitors of apoptosis (IAPs) are a family of proteins that regulate cell survival by binding to caspases to inhibit their catalytic activities [130]. The protein X-linked inhibitor of apoptosis (XIAP) is the most commonly expressed and the most potent endogenous caspase inhibitor among the IAPs, and it can be S-nitrosylated in several neurodegenerative disorders (Table 1). In fact, recent studies have shown a significant increase of S-nitrosylated XIAP in brain samples from PD, AD and Huntington's disease (HD) patient's possibly promoting apoptosis [131].
Transcription factors and cysteinet deregulation in AD
There are some examples of redox regulation of gene transcription in which cysteine residues form a fundamental part of the regulatory machinery (Figure 1). It has been shown that disulfide bridges control the formation of the homotrimer of the heat shock transcription factor 1 (HSF1) in mammals, which is translocated into the nucleus where it activates the transcription of both heat shock protein (Hsp) 70 and Hsp90 [132], which have been implicated in AD physiopathology. In oxidizing conditions, the intramolecular disulfide bond prevented the activation of HSF1, but under reducing conditions the intramolecular bond is broken and HSF1 cannot form the required trimer [132].
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a transcription factor that regulates the expression of antioxidant proteins protecting against oxidative damage. Nrf2 is kept in the cytoplasm by the Kelch like-ECH-associated protein 1 (Keap1) and Cullin-3, which degrade Nrf2 by ubiquitination [133]. Oxidative modification of critical cysteine residues in Keap1 disrupts the Keap1- Cullin-3 ubiquitination system, and Nrf2 is not ubiquitinated. The result is the translocation of Nrf2 into the nucleus where it combines with a small (musculoaponeurotic fibrosarcoma) Maf protein and binds to the antioxidant response element in the upstream promoter region of many antioxidant genes initiating their transcription [134].
N-Acetylcysteine (NAC) as A Natural Antidote for AD
The mechanisms of NAC actions, at the cellular level, are not completely understood. It has been argued that the principal mechanism explaining NAC beneficial actions is through the restoration of the glutathione pool and its redox capacity [135-137]. NAC does not need the alanine-serine-cysteine system to enter the cells since NAC is a membrane-permeable cysteine precursor that does not require active transport. Once into the cell, NAC is rapidly hydrolysed to yield cysteine [138], which can regenerate total glutathione content and reduce excessively oxidized glutathione levels. Another action of NAC is its direct scavenging ability against reactive species [139,140]. However, little attention has been focused on the effect of NAC on protein associated thiol regulation. NAC can restore the specific activities of mitochondrial complex I, IV and V in synaptic mitochondria obtained from aged mice, which was attributed to the restoration of thiol cysteine in those proteins. Likewise, in vivo experiments corroborate that the specific enzymatic activities of some of these complexes were restored by chronic NAC administration increasing ATP and GSH levels and decreasing lipid and protein oxidation in presynaptic terminals [136,141,142]. In the present paper, we have reviewed the functional consequences of redox modifications of different SCCPs that participate in a wide range of pathways implicated in AD physiopathology. In this context, NAC is a safety natural precursor of cysteine that can restore the redox environmental conditions for the adequate function of SCCPs. In fact, glutathione is a sensitive cysteine-containing tripeptide that depends on the cysteine/cystine ratio and the redox microenvironment.
Redox regulation of SCCPs may explain many effects of NAC including the improvement of metabolic function, calcium signaling, protein folding and turnover, which are closely associated with aging and AD development. However, modifications of cysteine residues in proteins by NAC have not been systematically studied yet. For example, NAC can alter the oxidative modifications of plasma proteins like transthyretin (TTR) by interacting with the sensitive cysteine residue of the protein in vitro and in vivo [142]. The interaction of NAC with TTR seems to be dose-dependent with a biphasic character of NAC-TTR interaction [142,143]. Indeed, TTR can suppress the AD phenotype in transgenic animal models, and it can reduce cerebral Aβ deposition [144]. Moreover, it has been shown that the administration of NAC can ameliorate the onset and severity of premature aging in Bmal1-deficient mice (Bmal1-/-mice). Bmal1 is a circadian clock protein implicated in tissue homeostasis by direct regulation of ROS, and it acts as a transcription factor of key components of the circadian clock. Administration of NAC attenuated the development of the age-associated phenotype of Bmal1-/- mice increasing the average and maximal lifespan of animals [145].
A little-explored mechanism of NAC is the redox regulation of gene transcription and expression pathways dependent on sensitive cysteine residues of transcription factors. Interestingly, NAC can directly modulate the activity of common transcription factors both in vitro and in vivo [146]. NAC treatment suppressed NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation in oxidative stressed cultured cells, and in clinical sepsis, reducing the subsequent cytokine production. NF-κB is naturally bound to its inhibitor (I-κB) that prevents its nuclear translocation. Dissociation of I-κB following its phosphorylation by the specific kinase of NF-κB (IKK) allows NF-κB transport to the nucleus [146]. In fact, it has been demonstrated that NAC down-regulates the transcription of the APP gene in neuroblastoma cells by decreasing the binding activity of the transcription factor NF-κB [147]. These effects of NAC are mediated through its ability to regulate and modulate the sensitive cysteine residues in those proteins that form part of the proposed cysteinet.
Genome-wide association studies have implicated numerous genes in AD [148] supporting the concept that altered APP misprocessing is only a part of the complex AD physiopathology. Therefore, diverse genetic alterations, risk factors and disturbed pathways may converge on the proposed cysteinet deregulation in AD, which may be prevented and restored by NAC. Taken together, NAC may protect neuronal cells working as an antioxidant but also regulating the oxidation of thiol groups of sensitive cysteine residues in multiple SCCPs, thereby restoring key cellular pathways that maintain neuronal survival. For example, it has been demonstrated that NAC activates the Ras-ERK (extracellular signal-regulated kinase) pathway in PC12 cells preventing neuronal death in the absence of trophic factors by a non-antioxidant mechanism [149]. Since Ras proteins have critical redox-sensitive cysteine residues [150], the authors suggested that NAC directly activates Ras through its reducing capacity [149]. Activation of the Ras-ERK pathway may also lead to the induction of c-fos and c-jun genes [149]. Indeed, NAC can induce c-fos and c-jun transcription in cellular systems [151]. Other studies have shown that NAC can protect human cortical neurons against apoptosis induced by Aβ-amyloid 1-42 [152] as well as inducing p35/Cdk5 pathway activation [153], and reducing phosphorylation/deactivation of MLK3-MKK7-JNK3 signaling cascade [154]. Therefore, in addition to antioxidant properties, and increasing GSH levels, NAC protects against Aβ toxicity through the activation of diverse signaling pathways. Finally, since down-regulation of peptidyl-prolyl isomerases 1 (Pin1) has been implicated in Alzheimer's disease [155] it was investigated the effect of chronic administration of NAC in human double mutant APP/PS-1 knock-mice. NAC treatment slightly increased the Pin1 levels, possibly decreasing Aβ induced oxidative stress [156].
Other probable beneficial action of NAC administration may be the prevention of age-related protein misfolding and aggregation through the regulation of cysteine oxidative modification associated with aging. The 3-D structures of proteins are related to their function, and conformational changes occur in many proteins when they accumulate in tissues [157]. Conformational changes are most likely to occur in proteins that have repetitive amino acid sequences, such as polyglutamine in HD. In this case, NAC has proven its beneficial effects in animal models [158]. Indeed, accumulation of misfolded proteins can contribute to the pathogenesis of age-related neurodegenerative diseases such as AD, PD and HD, which may be potentially restored by NAC administration. Indeed, NAC have shown beneficial effects in many different diseases including neurodegenerative diseases such as PD, and other brain injuries like cerebral ischemia and hemorrhage, traumatic brain injury, and psychiatric diseases [reviewed in 13,14].
Finally, sulfur compounds, particularly cysteine, inhibit the fibrillation of Aβ1-40 and Aβ1-42. Interestingly, aggregates of Aβ1-40 and Aβ1-42 induced by cysteine were less cytotoxic than those induced by catechin, which is the most typical inhibitor of amyloid fibril formation [159].
Preclinical and clinical studies of NAC in AD
AD is an age-associated neurodegenerative disease that has shown oxidative damage in the temporal and cerebral cortex and decreased glutathione concentrations in different cortical areas and the hippocampus [160-162]. Indeed, most clinical trials of antioxidants for the treatment of AD have employed α-tocopherol or selegiline, an irreversible and selective monoamine oxidase B (MAO-B) inhibitor in an attempt to counteract the pro-oxidant conditions of the disease. Besides, NAC has been tested in some murine models of AD [163,164], but studies in humans with neurodegenerative diseases are scarce.
Some preclinical studies have provided evidence that administration of NAC is beneficial in AD murine models counteracting oxidative damage [163-165] and decreasing Aβ1-40 and Aβ1-42 levels [166]. Besides, NAC was able to improve the results in the T-maze foot-shock avoidance paradigm [167]. NAC administration to human double mutant APP/PS-1 knock-mice before the deposition of Aβ in their brains, decreased protein and lipid oxidation, nitration of proteins, and increased glutathione peroxidase and reductase activities compared to age-matched controls [156]. Pretreatment with NAC in cultured neuroblastoma cells can also affect APP processing by influence both β-secretase and γ-secretase activities [168]. In the same study, NAC decreased phosphorylated tau levels in the presence or absence of stressing conditions (hydrogen peroxide and UV light exposition) through a pathway involving the modulation of phosphatases activities [168].
As previously mentioned, AD is associated with mitochondrial disorders including a reduced level of cytochrome c oxidase activity in post-mortem cerebral cortex. Additionally, ROS generation in cybrid cells transferred with mitochondria from AD platelets showed that complex IV defects are associated with the disease [169]. Besides, mitochondrial DNA mutations have also been demonstrated in AD patients [17-172], suggesting a role for mitochondrial DNA damage in the impairment of the oxidative phosphorylation and bioenergetic capacity associated with the disease [173]. Two studies have demonstrated a decrease in the mitochondrial mRNA that encodes complex IV in the temporal cortex and hippocampus of AD patients [174,175]. A study evaluated the effect of lipoic acid and NAC on oxidative and apoptotic markers in fibroblasts from patients with AD and age-matched and young controls. AD fibroblasts showed the highest levels of oxidative stress, and both antioxidants exerted a protective effect as evidenced by the decrease in oxidative stress and apoptotic markers. Moreover, the oxidative damage observed was associated with mitochondrial dysfunction, which was attenuated by both antioxidants. These data suggest that mitochondria must be a major target in AD therapies based on NAC supplementation [176]. Finally, we have demonstrated that chronic oral administration of NAC was able to restore cytochrome c oxidase activity measured in synaptic mitochondria from aged mice, decreasing the age-related oxidative damage in mitochondrial lipids and proteins [136,141,142]. This effect of chronic NAC administration was associated with a delay in age-associated memory impairment in aged animals [177].
In a study on probable AD patients, treatment with NAC for 24 weeks to drive cystine-glutamate antiporter system caused a beneficial trend in all measures tested as well as a significant improvement in some cognitive tests [178]. The release of glutamate from cystine-glutamate antiporter system and the subsequent activation of extra-synaptic NMDA (N-methyl-D-aspartate) receptors may contribute to β-amyloid production and toxicity [179,180]. In contrast, cystine uptake by cystine-glutamate antiporter system seems to exert beneficial effects by lowering β-amyloid stress, preventing apoptosis triggered by oxidative stress, and normalizing the activity of sodium-dependent glutamate transporters [168,181]. Another clinical study in a small group of patients with moderate to late-stage probable AD showed improvements in various neuropsychiatric tests after chronic administration of NAC in combination with folic acid, vitamin B12, α-tocopherol, S-adenosyl methionine and acetyl-L-carnitine [182]. A case report described a man with probable AD and hyperhomocysteinemia who showed significant clinical improvement after the administration of NAC, vitamin B12, and folinic acid [183].
Conclusions
Future studies are needed considering the essential role of cysteine residues for the maintenance of protein structure and function and the importance of NAC in various clinical and experimental setting. The present review emphasizes the singularity of NAC as the unique substance that, at present, is widely used in clinical practice, can cross the blood brain barrier (BBB) and can directly interact with key SCCPs into the brain counteracting brain aging and age-related neurodegenerative diseases. Indeed, recent preliminary results have demonstrated a beneficial effect of NAC on the dopamine system associated with the improvement of clinical parameters in PD [184]. In this study, patients received a DaTScan before and after treatment with NAC for 90 days, to measure dopamine transporter (DAT) binding. The study showed a significant increase in DAT binding in the caudate and putamen in the PD group treated with NAC without significant changes in the control group. In addition, the unified Parkinson's disease rating scale score, to measure clinical symptoms, were also significantly improved in the NAC group [184]. Therefore, differential cysteine labeling and global label-free proteomics studies in blood, CSF and brain tissue will be necessaries to understand the role of cysteinet deregulation in aging and age-associated neurodegenerative diseases. NAC can be potentially used to prevent and restore the deregulation of redox homeostasis through the replenishment of mitochondrial soluble (cysteine/glutathione) and protein-linked thiols, which may restore the mitochondrial bioenergetic capacity and biogenesis. Much remains to be investigated concerning redox signaling and modulation of the mitochondrial function, but it seems that redox regulation by NAC can play a key role in the cysteinet deregulation associated with aging, which is the principal risk factor for late-onset AD.
References
- Gouras GK,, Tampellini D, Takahashi RH, et al. (2010) Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol 119: 523-541.
- Wakabayashi K, Honer WG, Masliah E (1994) Synapse alterations in the hippocampal-entorhinal formation in Alzheimer's disease with and without Lewy body disease. Brain Res 667: 24-32.
- Lacor PN, Buniel MC, Furlow PW, et al. (2007) Abeta oligomer- induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 27: 796-807.
- Miller DL, Papayannopoulos IA, Styles J, et al. (1993) Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys 301: 41-52.
- Roher AE, Lowenson JD, Clarke S, et al. (1993) β.;-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA 90: 10836-10840.
- Citron M, Oltersdorf T, Haass C, et al. (1992) Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature 360: 672-674.
- Lesné S, Koh MT, Kotilinek L, et al. (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440: 352-357.
- Shankar GM, Li S, Mehta TH, et al. (2006) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14: 837-842.
- Grundke-Iqbal I, Iqbal K, Quinlan M, et al. (1986) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261: 6084-6089.
- Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA 83: 4044-4048.
- Filser S, Ovsepian SV, Masana M, et al. (2015) Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry 77: 729-739.
- Lambracht-Washington D, Rosenberg RN (2013) Advances in the development of vaccines for Alzheimer's disease. Discov Med 15: 319-326.
- Martinez-Banaclocha M (2016) Cysteine network (CYSTEINET) dysregulation in Parkinson's disease: role of N-acetylcysteine. Curr Drug Metab 17: 368-385.
- Martínez-Banaclocha M (2016) Cellular cysteine network (Cysteinet): pharmacological intervention in brain aging and neurodegenerative diseases. In: Frontiers in Clinical Drug Research-Central Nervous System; Atta-Ur-Rahman, Ed.; Bentham Science Publishers, (In press).
- Mailloux RJ, Jin X, Willmore WG (2014) Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol 2: 123-139.
- Paulsen CE, Carroll KS (2013) Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev 113: 4633-4679.
- Barja G (2002) Endogenous oxidative stress: relationship to aging, longevity and caloric restriction. Ageing Res Rev 1: 397-411.
- Bokov A, Chaudhuri A, Richardson A (2004) The role of oxidative damage and stress in aging. Mech Ageing Dev 125: 811-826.
- Perez VI, Bokov A, Van Remmen H, et al. (2009) Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790: 1005-1014.
- Jones DP, Mody Jr VC, Carlson JL, et al. (2002) Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Rad Biol Med 33: 1290-1300.
- Nuttall SL, Martin U, Sinclair AJ, et al. (1998) Glutathione: in sickness and in health. Lancet 351: 645-646.
- Liedhegner EAS, Gao XH, Mieyal JJ (2012) Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antiox Redox Sig 16: 543-566.
- Ansari MA, Scheff SW (2010) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69: 155-167.
- Go YM, Jones DP (2011) Cysteine/cystine redox signaling in cardiovascular disease. Free Rad Biol Med 50: 495-509.
- Miquel J, Ferrándiz ML, De Juan E, et al. (1995) N-acetylcysteine protecs against age-related decline of oxidative phosphorylation in liver mitochondria. Eur J Pharmacol 292: 333-332.
- McDonagh B, Sakellariou GK, Smith NT, et al. (2014) Differential cysteine labelling and global label-free proteomics reveals an altered metabolic state in skeletal muscle aging. J Proteome Res 13: 5008-5021.
- Freer R, Sormanni P, Vecchi G et al. (2016) A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer's disease. Sci Adv 2: e1600947.
- De Benedictis G, Carrieri G, Varcasia O, et al. (2000) Inherited variability of the mitochondrial genome and successful aging in humans. Ann New York Acad Sci 908: 208-218.
- Hamilton ML, van Remmen H, Drake JA, et al. (2001) Does oxidative damage to DNA increase with age? Proc Natl Acad Sci USA 98: 10469-10474.
- Yankner BA, Lu T, Loerch P (2008) The aging brain. Annu Rev Pathol 3: 41-66.
- Zahn JM, Poosala S, Owen AB, et al. (2007) AGEMAP: a gene expression database for aging in mice. PLoS Genet 3: e201.
- Bishop NA, Lu T, Yankner BA (2010) Neural mechanisms of ageing and cognitive decline. Nature 464: 529-535.
- Sohal RS, Forster MJ (2014) Caloric restriction and the aging process: a critique. Free Rad Biol Med 73: 366-382.
- Guo Z, Ersoz A, Butterfield DA, et al. (2000) Beneficial effects of dietary restriction on cerebral cortical synaptic terminals: preservation of glucose transport and mitochondrial function after exposure to amyloid β.;-peptide and oxidative and metabolic insults. J Neurochem 75: 314-320.
- Nakamura T, Tu S, Akhtar MW, et al. (2013) Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78: 596-614.
- Bouillaud F, Blachier F (2011) Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid Redox Signal 15: 379-391.
- Corder EH, Saunders AM, Strittmatter WJ, et al. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921-923.
- Heijmsns BT, Westendorp RG, Slagboom PE (2000) Common gene variants, mortality and extreme longevity in humans. Exp Gerontol 35: 865-877.
- Katzman R (1994) Apolipoprotein E and Alzheimer's disease. Curr Op Neurobiol 4: 703-707.
- Pedersen WA, Chan SL, Mattson MP (2000) A mechanism for the neuroprotective effect of apolipoprotein E: isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J Neurochem 74: 1426-1433.
- Abrams AJ, Farooq A, Wang G (2011) S-nitrosylation of ApoE in Alzheimer's disease. Biochemistry 50: 3405-3407.
- Reddie KG, Carroll KS (2008) Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol 12: 746-754.
- Fomenko DE, Xing W, Adair BM, et al. (2007) High-throughput identification of catalytic redox-active cysteine residues. Science 315: 387-389.
- Weerapana E, Wang C, Simon GM, et al. (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468: 790-795.
- D'Autreaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813-824.
- Levonen AL, Landar A, Ramachandran A, et al. (2004) Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defenses in response to electrophilic lipid oxidation products. Biochem J 378: 373-382.
- Akhtar MW, Sunico C R, Nakamura T, et al. (2012) Redox regulation of protein function via cysteine S-nitrosylation and its relevance to neurodegenerative diseases. Intern J Cell Biol 2012.
- Moreira PA, Carvalho C, Zhu X, et al. (2010) Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta 1802: 2-10.
- Swerdlow RH, Burns JM, Khan SM (2010) The Alzheimer's disease mitochondrial cascade hypothesis. J Alzh Dis 20: S265-S279.
- Chen JX, Yan SS (2010) Role of mitochondrial amyloid-β in Alzheimer's disease. J Alzh Dis 20: S569-S578.
- García-Escudero V, Martín-Maestro P, Perry G, et al. (2013) Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid Med Cell Long 2013: 162152.
- Dua H, Guoa L, Yana S, et al. (2010) Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA 107: 18670-18675.
- Wang L, Guo L, Lu L, et al. (2016) Synaptosomal Mitochondrial Dysfunction in 5xfad Mouse Model of Alzheimer's Disease. PLoS One 11: e0150441.
- Stauch KL, Purnell PR, Fox HS (2014) Quantitative proteomics of synaptic and nonsynaptic mitochondria:insights for synaptic mitochondrial vulnerability. J Proteome Res 13: 2620-2636.
- Francis BM, Yang J, Song BJ, et al. (2014) Reduced levels of mitochondrial complex I subunit NDUFB8 and linked complex I + III oxidoreductase activity in the TgCRND8 mouse model of Alzheimer's disease. J Alzh Dis 39: 347-355.
- Scott RA (1995) Functional significance of cytochrome c oxidase structure. Structure 3: 981-986.
- Martin CT, Scholes CP, Chan SI (1988) On the nature of cysteine coordination to CuA in cytochrome c oxidase. J Biol Chem 263: 8420-8429.
- Swerdlow RH, Kish SJ (2002) Mitochondria in Alzheimer's disease. Int Rev Neurobiol 53: 341-385.
- Cardoso SM, Proenca MT, Santos S, et al. (2004) Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging 25: 105-110.
- Parker WD, Mahr NJ, Filley CM, et al. (1994) Reduced platelet cytochrome c oxidase activity in Alzheimer's disease. Neurology 44: 1086-1090.
- Mutisya EM, Bowling AC, Beal MF (1994) Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem 63: 2179-2184.
- Bosetti F, Brizzi F, Barogi S, et al. (2002) Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging 23: 371-376.
- Devi L, Prabhu BM, Galati DF, et al. (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci 26: 9057-9068.
- Dragicevic N, Mamcarz M, Zhu Y, et al. (2010) Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzh Dis 20: S535-S550.
- Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125: 1241-1252.
- Nakamura T, Lipton SA (2011) Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Diff 18: 1478-1486.
- Chen H, Chan DC (2009) Mitochondrial dynamics fusion, fission, movement, and mitophagy in neurodegenerative diseases. Human Mol Genet 18: R169-R176.
- Shutt T, Geoffrion M, Milne R, et al. (2012) The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep 13: 909-915.
- Redpath CJ, Khalil MB, Drozdzal G, et al. (2013) Mitochondrial hyperfusion during oxidative stress is coupled to a dysregulation in calcium handling within a C2C12 cell model. PLoS One 8: e69165.
- Halestrap AP, Brenner C (2003) The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem 10: 1507-1525.
- Queiroga, CS, Almeida AS, Martel C, et al. (2010) Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J Biol Chem 285: 17077-17088.
- McStay GP, Clarke SJ, Halestrap AP. (2002) Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J 367: 541-548.
- McDonagh B, Martinez-Acedo P, Vazquez J (2012) Application of iTRAQ reagents to relatively quantify the reversible redox state of cysteine residues. Int J Proteomics 2012: 514847.
- Butterfield DA, Hardas SS, Bader Lange ML (2010) Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer disease: many pathways to neurodegeneration. J Alzh Dis 20: 369-393.
- Li S, Whorton AR (2003) Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch Biochem Biophys 410: 269-279.
- Burgoyne RD, Morgan A (2015) Cysteine string protein (CSP) and its role in preventing neurodegeneration. Sem Cell Develop Biol 40: 153-159.
- Tiwari SS, Troakes MDOC, Shurovi BN, et al. (2015) Evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer inverted question marks disease. Mol Brain 8: 6.
- Johnston PA, Südhof TC (1990) The multisubunit structure of synaptophysin. J Biol Chem 265: 8869-8873.
- Sze C, Troncoso JC, Kawas C et al. (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56: 933944.
- Hansen LA, Daniel SE, Wilcock GK, et al. (1998) Frontal cortical synaptophysin in Lewy body diseases: relation to Alzheimer's disease and dementia. J Neurol Neurosurg Psychiatry 64: 653-656.
- Forno LS (1996) Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol 55: 259-272.
- Jao CC, Der-Sarkissian A, Chen J, et al. (2004) Structure of membrane-bound α-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci USA 101: 8331-8336.
- Zhou W, Freed CR (2004) Tyrosine-to-cysteine modification of human α-synuclein enhances protein aggregation and cellular toxicity. J Biol Chem 279: 10128-10135.
- Stefanis L (2012) α-Synuclein in Parkinson's Disease. Cold Spring Harb Perspect Med 4:a009399.
- Südhof TC (2002) Synaptotagmins: Why So Many? J Biol Chem 277: 7629-7632.
- Levy-Lahad E, Wasco W, Poorkaj P, et al. (1995) Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269: 973-977.
- Sherrington R, Rogaev EI, Liang Y, et al. (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375: 754-760.
- Dimitrov M, Alattia J-R, Lemmin T, et al. (2013) Alzheimer's disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat Commun 4: 2246.
- Pike CJ, Burdick D, Walencewicz AJ, et al. (1993) Neurodegeneration induced by β-amyloid peptides in vitro: The role of peptide assembly state. J Neurosci 13:1676-1687.
- Laudon H, Hansson EM, Melen K, et al. (2005) A nine transmembrane domain topology for presenilin 1. J Biol Chem 25: 25.
- Kornilova AY, Kim J, Laudon H, et al. (2006) Deducing the transmembrane domain organization of presenilin-1 in γ-secretase by cysteine disulfide crosslinking. Biochemistry 45: 7598-7604.
- Tedde A, Nacmias B, Ciantelli M, et al. (2003) Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch Neurol 60: 1541-1544.
- Tolia A, Chavez-Gutierrez L, Strooper BD (2006) Contribution of Presenilin Transmembrane Domains 6 and 7 to a Water-containing Cavity in the γ-Secretase Complex. J Biol Chem 281: 27633-27642.
- Payne AJ, Gerdes BC, Naumchuk Y, et al. (2013) Presenilins regulate the cellular activity of ryanodine receptors differentially through isotype-specific N-terminal cysteines. Exp Neurol 250: 143-150.
- Bhattacharyya R, Barren C, Kovacs DM (2013) Palmitoylation of Amyloid Precursor Protein Regulates Amyloidogenic Processing in Lipid Rafts. J Neurosci 33: 11169 -11183.
- Barnham KJ, McKinstry WJ, Multhaup G, et al. (2003) Structure of the Alzheimer's disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem 278: 17401-17407.
- Schweers O, Mandelkow E, Biernat J, et al. (1995) Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein T controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci USA 92: 8463-8467.
- Landino LM, Skreslet TE, Alston JA (2004) Cysteine oxidation of Tau and microtubule-associated protein-2 by peroxynitrite. J Biol Chem 279: 35101-35105.
- Cohen TJ, Friedmann D, Hwang AW, et al. (2013) The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat Struct Mol Biol 20: 756-762.
- Wegmann S, Medalsy ID, Mandelkow E, et al. (2012) The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectric brush. Proc Natl Acad Sci USA 109: E313-321.
- Soeda Y, Yoshikawa M, Almeida OF, et al. (2015) Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat Commun 6: 10216.
- Thibault O, Gant JC, Landfield PW (2007) Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell 6: 307-317.
- Berridge MJ (2013) Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 7: 2-13.
- Stutzmann GE, Smith I, Caccamo A, et al. (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci 26: 5180-5189.
- Sun J, Xu L, Eu JP, et al. (2003) Nitric oxide, NOC-12, and S-nitrosoglutathione modulate the skeletal muscle calcium release channel/ryanodine receptor by different mechanisms. An allosteric function for O2 in S-nitrosylation of the channel. J Biol Chem 278: 8184-8189.
- Bánsághi S, Golenár T, Madesh M, et al. (2014) Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem 289: 8170-8181.
- Khan SA, Rossi AM, Riley AM, et al. (2013) Subtype-selective regulation of IP3 receptors by thimerosal via cysteine residues within the IP3-binding core and suppressor domain. Biochem J 451: 177-184.
- Kurumatani T, Fastboma J, Bonkalea WL, et al. (1998) Loss of inositol 1,4,5-trisphosphate receptor sites and decreased PKC levels correlate with staging of Alzheimer's disease neurofibrillary pathology. Brain Research 796: 209-221.
- Tonga XY, Yinga J, Pimentel DR, et al. (2008) High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol 44: 361-369.
- Sharov VS, Dremina ES, Galeva NA (2006) Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J 394: 605-615.
- Qina M, Wanga W, Thirumalai D (2015) Protein folding guides disulfide bond formation. Proc Natl Acad Sci USA 112: 11241-11246.
- Haase-Pettingell C, Betts S, Raso SW, et al. (2001) Role for cysteine residues in the in vivo folding and assembly of the phage P22 tail spike. Protein Sci 10: 397-410.
- Winklhofer KF, Tatzeltand J, Haass C (2008) The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J 27: 336-349.
- Shin H-C (2001) Protein Folding, misfolding, and refolding of therapeutic proteins. Biotechnol Bioprocess Eng 6: 237-243.
- Creighton TE (1986) Disulfide bonds as probes of protein folding pathways. Methods Enzymol 131: 83-106.
- Uehara T, Nakamura T, Yao D, et al. (2006) S-Nitrosylated protein disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513-517.
- Riemer J, Bulleid N, Herrmann JM (2009) Disulfide formation in the ER and mitochondria: two solutions to a common process. Science 324: 1284-1287.
- Koch JR, Schmid FX (2014) Mia40 targets cysteines in a hydrophobic environment to direct oxidative protein folding in the mitochondria. Nat Comm 5: 3041.
- Garcia-Mata R, Gao YS, Sztul E (2002) Hassles with taking out the garbage: aggravating aggresomes. Traffic 3: 388-396.
- Lecke, SH, Goldberg AL, Mitch WE (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807-1819.
- Jahngen-Hodge J; Obin MS, Gong X, et al. (1997) Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem 272: 28218-28226.
- Doris KS, Rumsby EL, Gong BA (2012) Oxidative stress responses involve oxidation of a conserved ubiquitin pathway enzyme. Mol Cell Biol 32: 4472-4481.
- Hong L, Huang HC, Jiang ZF (2014) Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer's disease. Neurol Res 36: 276-282.
- Fischer DF, van Dijk R, van Tijn P, et al. (2009) Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol Aging 30: 847-863.
- Hasanbasic S, JahicA, Karahmet E (2016) The role of cysteine proteases in Alzheimer disease. Mater Sociomed 28: 235-238.
- Trinchese F, Fa' M, Liu S, et al. (2008). Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest 118: 2796-2807.
- Moldoveanu T, Hosfield CM, Lim D, et al. (2002) A Ca(2+) switch aligns the active site of calpain. Cell 108: 649-660.
- Ono Y, Sorimachi H (2012) Calpains-An elaborate proteolytic system. Biochim Biophys Acta 1824: 224-236.
- Melino G, Bernassola F, Knight RA, et al. (1997) S-nitrosylation regulates apoptosis. Nature 388: 432-433.
- Salvesen GS, Duckett CS (2002) IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 3: 401-410.
- Tsang AHK, Lee YIL, Ko HS et al. (2009) S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc Nat Acad Sci USA 106: 4900-4905.
- Lu M, Kim HE, Li CR, et al. (2008) Two distinct disulfide bonds formed in human heat shock transcription factor 1 actin opposition to regulate its DNA binding activity. Biochemistry 47: 6007-6015.
- Sekhar KR,Yan XX, Freeman ML (2002) Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene 21: 6829-6834.
- Piccirillo S, Filomeni G, Brune B, et al. (2009) Redox mechanisms involved in the selective activation of Nrf2-mediated resistance versus p53-dependent apoptosis in adenocarcinoma gastric cells. J Biol Chem 284: 27721-27733.
- Meister A (1988) Glutathione metabolism and its selective modification. J Biol Chem 263: 17205-17208.
- Martínez-Banaclocha M (2001) Therapeutic potential of N-acetyl-cysteine in age-related mitochondrial neurodegenerative diseases. Med Hypoth 56: 472-477.
- Aoyama K, Suh SW, Hamby AM, et al. (2006) Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci 9: 119-126.
- Sen O, Caner H, Aydin MV, et al. (2006) The effect of mexiletine on the level of lipid peroxidation and apoptosis of endothelium following experimental subarachnoid hemorrhage. Neurol Res 28: 859-863.
- Kerksick C, Willoughby D (2005) The Antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J Int Soc Sports Nutr 2: 38-44.
- Medved I, Brown MJ, Bjorksten AR, et al. (2003) N-acetylcysteine infusion alters blood redox status but not time to fatigue during intense exercise in humans. J Appl Physiol 94: 1572-1582.
- Martínez-Banaclocha M (2000) N-acetylcysteine increase in complex I activity in synaptic mitochondria from aged mice: implications for treatment of Parkinson's disease. Brain Res 859: 173-175.
- Martínez-Banaclocha M, Martínez N (1999) N-acetylcysteine elicited increase in cytochrome c oxidase activity in mice synaptic mitochondria. Brain Res 842: 249-251.
- Henze A, Raila J, Scholze A, et al. (2013) Does N-acetylcysteine modulate post-translational modifications of transthyretin in hemodialysis patients? Antiox Redox Sign 19: 1166-1672.
- Li X, Zhang X, Ladiwala AR, et al. (2013) Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J Neurosci 33: 19423-19433.
- Kondratov RV, Vykhovanets O, Kondratova AA, et al. (2009) Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging 1: 979-987.
- Samuni Y, Goldstein S, Dean OM, et al. (2013) The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta 1830: 4117-4129.
- Studer R, Baysang G, Brack C (2001) N-acetyl-L-cystein downregulates beta-amyloid precursor protein gene transcription in human neuroblastoma cells. Biogerontology 2: 55-60.
- Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45: 1452-1458.
- Yan CY, Greene LA (1998) Prevention of PC12 cell death by N-Acetylcysteine requires activation of the Ras pathway. J Neurosci 18: 4042-4049.
- Hobbs GA, Gunawardena HP, Campbell SL (2014) Biophysical and proteomic characterization strategies for cysteine modifications in Ras GTPases. Meth Mol Biol 1120: 75-96.
- Li W, Wang G, Wang R, et al. (1994) The redox active components H2O2 and N-acetyl-L-cysteine regulate expression of c-jun and c-fos in lens systems. Exp Eye Res 59: 179-190.
- Medina S, Martinez-Banaclocha M, Hernanz A (2002) Antioxidants inhibit the human cortical neuron apoptosis induced by hydrogen peroxide, tumor necrosis factor alpha, dopamine and beta-amyloid peptide 1-42. Free Radic Res 36: 1179-1184.
- Hsiao YH, Chen PS, Yeh SH, et al. (2008) N-acetylcysteine prevents beta-amyloid toxicity by a stimulatory effect on p35/cyclin-dependent kinase 5 activity in cultured cortical neurons. J Neurosci Res 86: 2685-2695.
- Xu Y, Hou XY, Liu Y, et al. (2009) Different protection of K252a and N-acetyl-L-cysteine against amyloid-beta peptide-induced cortical neuron apoptosis involving inhibition of MLK3-MKK7-JNK3 signal cascades. J Neurosci Res 87: 918-927.
- Sultana R, Boyd-Kimball D, Poon HF, et al. (2006) Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus: A redox proteomics analysis. Neurobiol Aging 27: 918-925.
- Huang Q, Aluise CD, Joshi G, et al. (2010) Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: toward therapeutic modulation of mild cognitive impairment. J Neurosci Res 88: 2618-2629.
- Reynaud E (2010) Protein misfolding and degenerative diseases. Nat Edu 3: 28.
- Wright DJ, Renoir T, Smith ZM et al. (2015) Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington's disease. Transl Psychiat 5: e492.
- Takai E, Uda K, Yoshida T, et al. (2014) Cysteine inhibits the fibrillation and cytotoxicity of amyloid-β 40 and 42: implications for the contribution of the thiophilic interaction. Phys Chem Chem Phys 16: 3566-3572.
- Adams JD Jr, KlaidmanLK, Odunze IN, et al. (1991) Alzheimer's and Parkinson's disease. Brain levels of glutathione, glutathione disulfide, and vitamin E. Mol Chem Neuropathol 14: 213-226.
- Jenner P (1994) Oxidative damage in neurodegenerative disease. Lancet 344: 796-798.
- Lohr JB, Browning JA (1995) Free radical involvement in neuropsychiatric illness. Psychopharmacol Bull 31: 159-165.
- Tchantchou F, Graves M, Rogers E, et al. (2005) N-acteylcysteine alleviates oxidative damage to central nervous system of ApoE-deficient mice following folate and vitamin E-deficiency. J Alzh Dis 7: 135-138.
- Tucker S, Ahl M, Bush A, et al. (2005) Pilot study of the reducing effect on amyloidosis in vivo by three FDA pre-approved drugs via the Alzheimer's APP 5′ untranslated region. Curr Alzh Res 2: 249-254.
- Tchantchou F, Graves M, Ortiz D, et al. (2004) Dietary supplementation with 3-deaza adenosine, N-acetyl cysteine, and S-adenosyl methionine provide neuroprotection against multiple consequences of vitamin deficiency and oxidative challenge. Neuromol Med 6: 93-103.
- Tucker S, Ahl M, Cho HH, et al. (2006) RNA therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin, and N-acetyl cysteine. Curr Alzh Res 3: 221-227.
- Farr SA, Poon HF, Dogrukol-Ak D, et al. (2003) The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem 84: 1173-1183.
- Olivieri G, Baysang G, Meier F, et al. (2001) N-Acetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: effects on beta-amyloid secretion and tau phosphorylation. J Neurochem 76: 224-233.
- Swerdlow BJ, Parks JK, Cassarino SW, et al. (1997) Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology 49: 918-925.
- Blanchard BJ, Park T, Fripp WJ, et al. (1993) A mitochondrial DNA deletion in normaly aging and in Alzheimer brain tissue. Neuroreport 4: 799-802.
- Hutchin T, Cortopassi G (1995) A mitochondrial DNA clone is associated with increased risk for Alzheimer's disease. Proc Natl Acad Sci USA 92: 6892-6895.
- Soffner JM, Brawn MD, Torroni A, et al. (1993) Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 17: 171-184.
- Beal MF (1996) Mitochondria, free radicals, and neurodegeneration. Curr Op Neurobiol 6: 661-666.
- Chandrasekaran K, Giordano T, Brady DR, et al. (1994) Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer's disease. Brain Res Mol Brain Res 24: 336-340.
- Simonian NA, Hyman BT (1994) Functional alterations in Alzheimer's disease: selective loss of mitochondrial-encoded cytochrome oxidase mRNA in the hyppocampal formation. J Neuropathol Exp Neurol 53: 508-512.
- Moreira PI, Harris PL, Zhu X, et al. (2007) Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J Alzh Dis 12: 195-206.
- Martínez-Banaclocha M, Hernández AI, Martínez N (2000) N-acetylcysteine delays age-associated memory impairment in mice: role in synaptic mitochondria. Brain Res 855: 100-106.
- Adair JC, Knoefel JE, Morgan N (2001) Controlled trial of N-acetylcysteine for patients with probable Alzheimer's disease. Neurology 57: 1515-1517.
- Qin S, Colin C, Hinners I, et al. (2006) System xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci 26: 3345-3356.
- Bordji K, Becerril-Ortega J, Nicole O, et al. (2010) Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-β production. J Neurosci 30: 15927-15942.
- Tanel A, Averill-Bates DA (2007) Inhibition of acrolein-induced apoptosis by the antioxidant N-acetylcysteine. J Pharmacol Exp Ther 321: 73-83.
- Remington R, Chan A, Paskavitz J, et al. (2009) Efficacy of a vitamin/nutriceutical formulation for moderate-stage to later-stage Alzheimer's disease: a placebo-controlled pilot study. Am J Alzheimers Dis Other Dement 24: 27-33.
- McCaddon A, Davies G (2005) Co-administration of N-acetylcysteine, vitamin B12 and folate in cognitively impaired hyperhomocysteinaemic patients. Int J Geriatr Psychiatry 20: 998-1000.
- Monti DA, Zabrecky G, Kremens D, et al. (2016) N-Acetyl cysteine may support dopamine neurons in Parkinson's disease: preliminary clinical and cell line data. PLoS One 11: e0157602.
Corresponding Author
Marcos Martínez-Banaclocha, Pathology Service at the Lluis Alcanyis Hospital, Xátiva, Valencia, Spain.
Copyright
© 2016 Martínez-Banaclocha M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.