Stress-Induced Down-Regulation of Nitric Oxide Synthase in the Rat Heart
Abstract
Objectives: beta3-adrenoceptor (beta3-AR) is expressed in a smaller proportion than beta1-AR and beta2-AR in the hearts of rats and humans and is activated in the presence of high concentrations of catecholamines. In the myocardium, beta3-ARs couple to G inhibitory protein that activates nitric oxide synthases (NOS), thus increasing nitric oxide production that reduces inotropism. Beta2-ARs and beta1-ARs remodeling was described in failing and senescent human and rodent hearts as well as in the heart of foot shock stressed rats. This report aims to evaluate the function and expression of beta3-AR as well as the expression of NOS isoforms in the heart of foot shock stressed rats.
Methods: Male Wistarrats (Rattusnorvegicus; 250-350 g; 12 weeks-old) were submitted to a 30 min session of foot shock stress (1 mA, 1s, 15-25 s interval between pulses) during 3 days and were euthanized immediately after the third stress session. The heart was isolated and dose-response curves to the beta3-AR selective agonist, BRL37, 344, was determined in the right atria, in the absence and in the presence of L-NAME. mRNA and protein expressions of beta3-AR and nitric oxide synthase (NOS) isoforms were analyzed in the four heart chambers of control and stressed rats.
Results: Atria sensitivity to BRL 37, 344 in the presence or absence of L-NAME was not altered. Beta3-AR mRNA and protein expressions were similar in the four heart chambers of control and stressed rats; mRNA and protein expressions of endothelial NOS (eNOS) and inducible NOS (iNOS), as well as eNOS phosphorylation were reduced by stress.
Conclusion: Environmental stress may alter the expression of proteins involved in the beta-3-AR signaling pathway. If the stress-induced reduction on eNOS and iNOS expressions would impair the cardio protective mechanisms and the systolic and diastolic functions remains to be investigated.
Keywords
Stress, Heart, Nitric oxide synthase, Catecholamines, β3-adrenergic receptors
Introduction
The mammalian heart function is under the control of the sympathetic and parasympathetic nervous system. Whereas vagal stimulation exerts negative effects on inotropism, sympathetic stimulation leads to an increase of heart rate and force of contraction due to the action of noradrenaline and adrenaline on β-adrenoceptors (β-AR) [1,2].
The three subtypes of β-ARs are expressed in the hearts of humans and rodents. β1-adrenoceptors (β1-AR) predominate over β2-adrenoceptors (β2-AR) in a ratio of about 80/20 [1-3] while the expression of β3-adrenoceptors (β3-AR) is considerably lower [4-6]. Additionally, because cardiac β3-ARs are activated only in the presence of high concentrations of catecholamines [5-7], they are predominantly inactive during normal physiologic conditions [8]. Nevertheless, their activation attenuates the response to intense adrenergic stimulation [9,10].
β-ARs belong to the class of G-protein coupled receptors. Upon activation, β1-AR and β2-AR couple to G stimulatory (Gs) protein-adenylyl cyclase (AC)-cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) signaling pathway. Targets of PKA include L-type calcium channels, T troponin and phospholamban [5,11,12]. These proteins phosphorylation contributes to cardiomyocytes' increase in beating rate, developed tension, and velocity of relaxation, thus increasing cardiac output. β2-AR may also couple to G inhibitory (Gi) protein, with opposite effects on AC to those of Gs stimulation. β2-AR-Gi also activates the phosphatidylinositol 3-kinase (PI3K)-AKT signaling pathway that controls cardiomyocytes life and death [13].
The third β-AR expressed in the myocardium, β3-AR, couple to Gi/o protein [14], stimulates guanylate cyclase that generates cyclic guanosine monophosphate (cGMP) resulting in activation of protein kinase G (PKG) that activates nitric oxide synthases (NOS). Three isoforms of NOS are expressed in the healthy myocardium. Endothelial NOS (eNOs) is found in the caveolae whereas inducible NOS (iNOS) and neuronal NOS (nNOS) are found predominantly in the cytosol. In contrast with constitutive eNOS and nNOS, iNOS is expressed in the myocardium in response to hypoxia, oxidative stress, or inflammatory cytokines [15]. The nitric oxide (NO) produced by the action of NOS has ubiquitous effects including reducing inotropism [14] that counterbalance the effects of β1- β2-AR-Gs over activation. In cardiac pathologies as, for example, in failing heart [16,17], and senescence [18], which have been associated with reduction in the β1-AR/β2-AR ratio [19-21],the β3-AR expression is increased and the NO synthesized by the action of NOS seems to have beneficial effects [19].
The NO effects are accomplished via post-translational S-nitrosylation of thiol groups of specific cysteine residues [22], depending on the redox environment [23]. Some substrates of S-nitrosylation include proteins involved in the excitation-contraction coupling process [24,25], notably troponin-C, phospholamban and sodium-calcium exchanger (NCX) 1. S-nitrosylation of the calcium-cycling machinery and the basal activity of the nNOS isoform are required for normal cardiac function [26].
As previously reported, in the heart of rats submitted to foot shock stress there is a reduction of β1-AR/β2-AR ratio [26]. As a consequence of this β-AR population remodeling, cardiac chronotropic and inotropic responses to catecholamines are altered [26]. The behavior of β3-AR and its downstream target, NOS, in the heart of rats where β1-AR and β2-AR expressions were altered, has not been investigated yet. The aim of this report was to evaluate the β3-AR function and expression as well as NOS isoforms expression in the heart of foot shock stressed rats.
Material and Methods
Animals
Male Wistarrats (Rattusnorvegicus; 250-350 g; 12 weeks-old), obtained from "Center for the Development of Experimental Models" (CEDEME), Federal University of São Paulo (São Paulo, SP, Brazil), were housed in standard cages in a temperature-controlled room (22 ℃) on a 12:12 h light: Dark cycle, with the lights on at 7: 00 am. Standard laboratory chow and tap water were available ad libitum. The experimental protocols were approved by the Ethic Committee on Animal Use of the Federal University of São Paulo (CEUA/UNIFESP), protocol number 1835/09, in accordance with National Council for Control of Animal Experimentation (CONCEA, Brazil).
Stress protocol
The foot shock stress protocol has been used by our group, as previously described [26,27]. The rats were randomly distributed in two groups: control (CO) and stress (ST). The rats of the stress group were placed in a Plexiglas chamber (26 × 21 × 26 cm) provided with a grid floor made of stainless-steel rods (0.3 cm in diameter, spaced 1.5 cm apart). During the 30 min sessions, which occurred between 7:30 am and 11:00 am, on three consecutive days, the foot shocks were delivered by a constant current source controlled by a microprocessor-based instrument. The current intensity was 1.0 mA, duration 1.0 s with pulses delivered at random intervals of 5-25 s. The rats were returned to the standard cages at the end of the first and the second foot shock sessions. After the third session, the rats were euthanized and hearts were harvested. The rats in the control group were placed in a similar Plexiglas chamber during 30 min, once a day for three days, between 7:30 a.m. and 11:00 a.m. However, they did not receive foot shocks.
Organ-bathstudies
The studies with isolated atrium were made in organ-bath, as previously described [26]. Right and left atrium were isolated and suspended in 20mL organ baths containing Krebs-Henseleit solution (in mmol/L: NaCl 115.0, KCl 4.6, CaCl2·2H2O 2.5, KH2PO4 1.2, MgSO4·7H2O 2.5, NaHCO3 25, glucose 11.0, and ascorbic acid 0.11). The solution was warmed at 36.5 ± 0.1 ℃ and continuously gassed with 95 % O2 and 5 % CO2 (pH 7.2-7.4). Developed tension was recorded by Power Lab amplifiers on Lab Chart software (version 7.3; AD Instruments, Castle Hill, New South Wales, Australia). Right and left atria were submitted to a resting tension of 4.9mN and remained in the bath for 45 min to stabilize the frequency of beats (chronotropism) and developed tension (inotropism), respectively.
Both atria were attached to isometric force transducers (Panlab, Cornella, Spain). Right atrium was submitted to a tension sufficient to allow recording of the spontaneous beating due to pacemaker activity, and the evaluation of the chronotropic effect of β-AR agonists. Left atrium was set under a resting tension of 4.9 mN and was electrically stimulated with pulses in a constant frequency of 2 Hz, 5 ms duration, and intensity 20% higher than the threshold. Pulses were delivered by a bipolar platinum electrode connected to a Grass S88 stimulator (Grass Instruments, Quincy, Massachusetts, USA). Pulses intensity was directly proportional to the developed tension so that the inotropic effect of β-AR agonists could be estimated. The developed tension was recorded by Power Lab amplifiers on Lab Chart software for Windows version 7.3 (AD Instruments, Castle Hill, New South Wales, Australia). A length-force curve was determined and each atrium length set to obtain a resting tension that yielded 70 % -80 % of the maximum developed force. The tension developed by the atrium in each contraction under this condition and in the absence of any agonist is referred to as the basal tension.
Cumulative concentration-response curves to BRL 37, 344 ((RRþSS)-(7)-4-[2-(2-(3-chlorophenyl)-2-hydroxyethyl) amino) propyl] phenoxy-acetate), a β3-AR agonist, were obtained in the presence or absence of L-NAME (NOS inhibitor; 10μM). The maximum response was determined when three crescent doses of the agonist did not change the response. Inotropic response was expressed in mili-Newtons (mN) of tension developed by 100mg of wet tissue (mN/100 mg). Chronotropic response was presented in beats per minute (beat/min). The sensitivity to the agonist was evaluated by determining the concentration of agonist that produced 50 % of the maximum response (EC50), and it was expressed as pD2 value, i.e., the negative logarithm of the EC50.
RT-PCR
One fragment of no more than 100 mg of right and left atria and ventricles was individually homogenized in 1mL TRIzol (Invitrogen Carlsbad, CA, EUA) using an T-18 Ultra-Turrax homogenizer (Ika Works Inc., Wilmington, NC, EUA) in an ice bath. Then, 200 μl of chloroform were added, vortex for 15 s, and rest for 3 min at room temperature. After that, the sample was centrifuged at 12,000 g, 15 min, 4 ℃. The water phase was transferred to a clean tube. Then, 500 μl of isopropanol was added and incubated during 10 min at room temperature. After that, the tube was centrifuged at 7, 500 g during 10 min at 4 ℃. The pellet was washed with 1 ml ethanol at 100%, resuspended in 30 μl of water treated with 0.1 % dietilpyrocarbonate (DEPC Ultra Pure, Invitrogen, Carlsbad, CA, EUA), and stored at - 80 ℃ until the moment of the assay. Then, the sample was thawed at room temperature, mRNA concentration and degree of purity were determined in Nanodrop 2000 c (Thermo Scientific, Waltham, MA, USA) under 260/280 nm. The mRNA was treated with DNAse (Deoxyribonuclease Amp Grade I, Invitrogen, Carlsbad, CA, EUA), according to the manufacturer indication. The final volume was of 10 μL. After mRNA purification, complementary DNA (cDNA) was obtained, using the High-Capacity cDNA Reverser Transcription kit (Applied Biosystems, Carlsbad, CA, USA), according to the manufacture indication in a final volume of 20 μL. The gene expression was analyzed using previously designed primers. Glyceraldehyde triphosphate dehydrogenase (GAPDH) was used as endogenous control. Real time-PCR was done using SYBRGreen PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) in a Step One Plus Real Time PCR System (Applied Biosystems, Carlsbad, CA, USA). The results were obtained by the 2-ΔΔCt method and were normalized in relation to the values obtained for the endogenous gene.
Western blot assay
The frozen right and left atria and ventricles were individually homogenized using a T-18 Ultra-Turrax homogenizer (Ika Works Inc., Wilmington, NC, USA) in ice-cold assay buffer (4 ℃) (0.1mM Pipes, 5mM MgCl2, 5mM EDTA, 1% Triton X100 (v/v, Bio Rad Laboratories Inc., Hercules, CA, USA), 20% glycerol (v/v) and cocktail inhibitors protease (Complete, Roche Applied Science, Perzberg, Upper Bavaria, Germany). The samples were centrifuged for 30 min at 10,000 g and the supernatant was collected and assayed for total protein concentration using the Bradford method (Bio Rad Laboratories Inc., Hercules, CA, USA). Samples were stored at - 80 ℃ until assay. Two hundred micrograms of total protein were separated using SDS-polyacrylamide gel electrophoresis (Mini-Protean III, Bio-Rad Inc., Hercules, CA, USA) and transferred to nitrocellulose membranes (Amersham Biosciences, UK). Membranes were blocked with 5 % non-fat milk in buffer (10 mM Tris-HCl (pH 7.6), 150mM NaCl and Tween 20 (0.02%, w/v) during 1 h. The following primary antibodies (from Santa Cruz Biotechnology, Dallas, USA) were incubated at 4 ℃ overnight with 1:1000 dilution in the same buffer with 3 % non-fat milk: β3-AR rabbitpolyclonal (sc1473); eNOS rabbitpolyclonal (sc654); iNOS rabbitpolyclonal (sc651); nNOS rabbitpolyclonal (sc648) and GAPDH rabbitpolyclonal(sc25778). The membranes were subsequently rinsed three times (5 min each) in buffer solution and then incubated with 2 µCi de 125I -protein marked (30 µCi/µg) for 2 hours at room temperature, followed by 30 min of washing. The membranes were exposed to the respective HRP-conjugated secondary antibody 1:2000 dilution (Santa Cruz Biotechnology, Dallas, USA) for 1 h and exposed to Kodak O-OMAT-AR photographic film (Kodak, Rochester, NY, USA). Densitometric analyses were done using Image J Launcher software.
Statistical analysis
The results were expressed as means ± s.e.m. Student's unpaired t-test was used to compare two groups (control versus stress). Analysis of variance (ANOVA) followed by Tukey test was used for multiple comparisons. Differences were considered significant at p ≤ 0.05. All statistical analyses were done using Prism v.8 (GraphPad Software Inc., San Diego, CA, USA).
Results
β3-adrenoceptor function and expression are not altered by foot shock stress
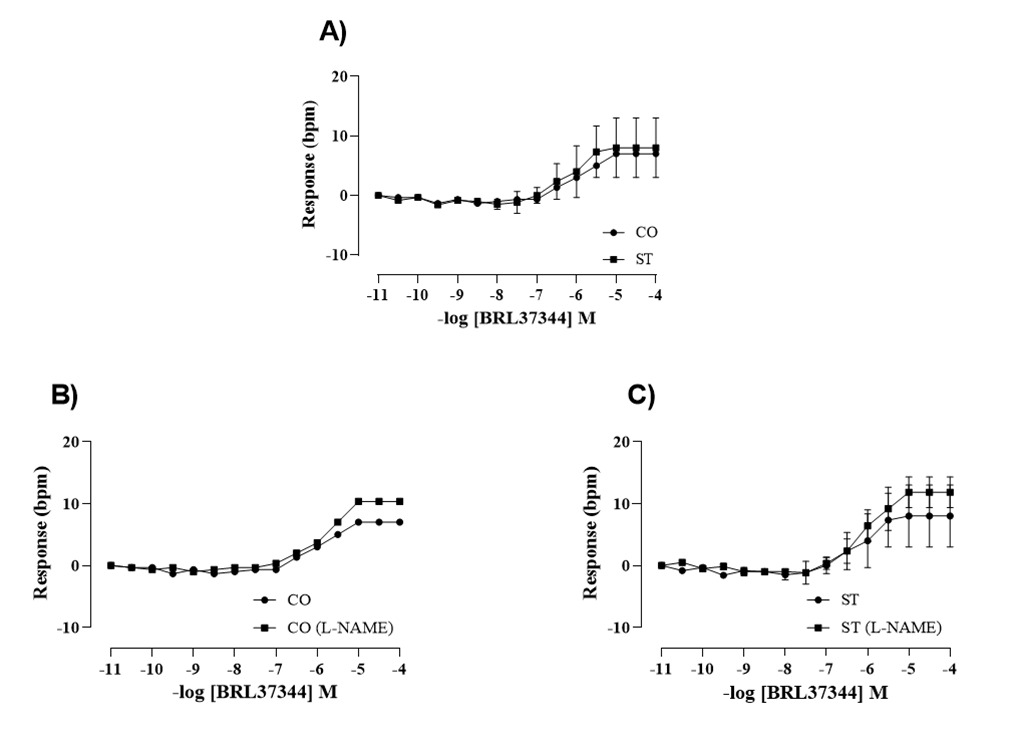
Figure 1A shows the concentration-response curves to BRL 37, 344 obtained in right atria isolated from control and stressed rats. BRL 37, 344 in the range of nanomolar concentrations had no effect in the beating rate of right atria of control or stressed rats. Positive chronotropic effect of BRL 37, 344 can be seen in the range of micromolar concentration. At this concentration, BRL 37, 344 is no longer selective for β3-AR, and this response is due to BRL 37, 344 coupling to β1-AR [5]. Figures 1B and 1C show the concentration-response curves to BRL 37, 344 in the absence and in the presence of L-NAME in the right atria of control and stressed groups, respectively. The sensitivity and maximum response to BRL 37, 344 were not altered by stress and by L-NAME.
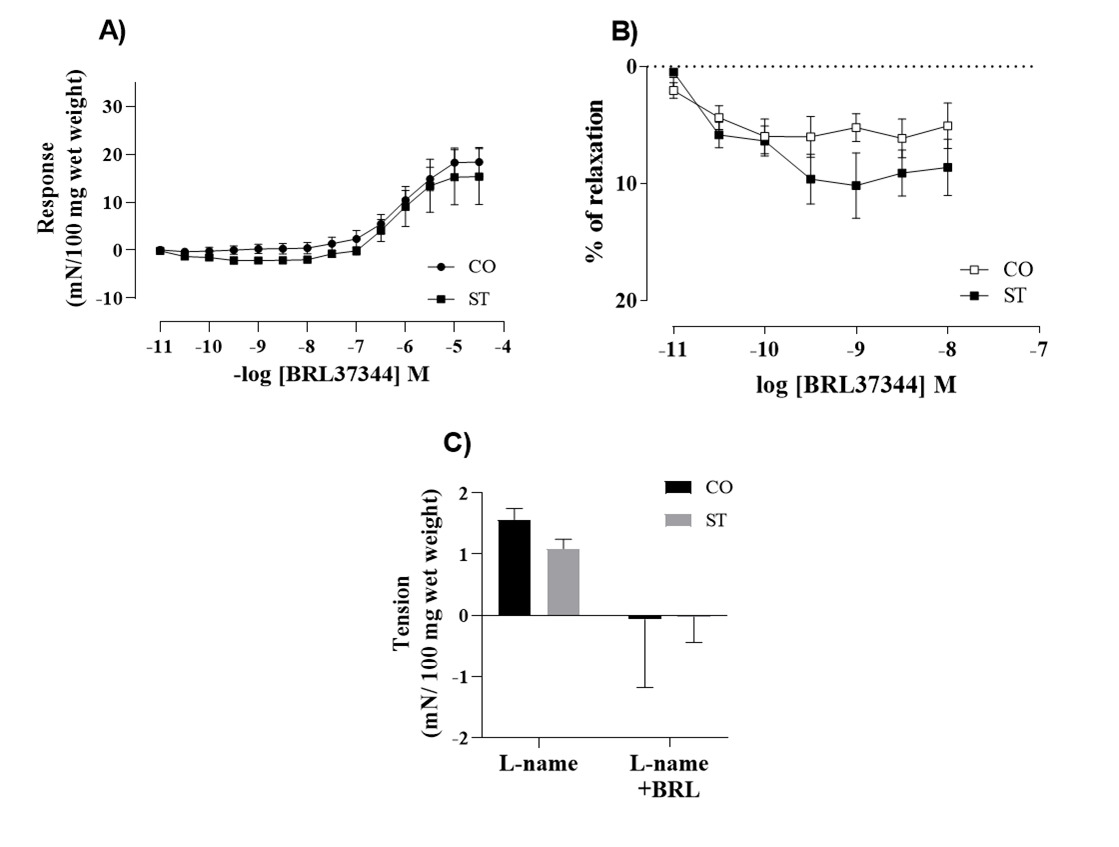
The inotropic response to BRL 37, 344 was evaluated in the left atria and it is shown in the Figure 2. A positive inotropic response was seen in the range of micromolar concentrations that did not differ between groups (Figure 2A). Figure 2B shows that in the range of nanomolar concentration BRL 37, 344 induced a negative inotropic effect in the left atria of stressed rats. The response of left atria to a single 10 nM dose of BRL 37, 344 was abolished by L-NAME in both groups (Figure 2C).
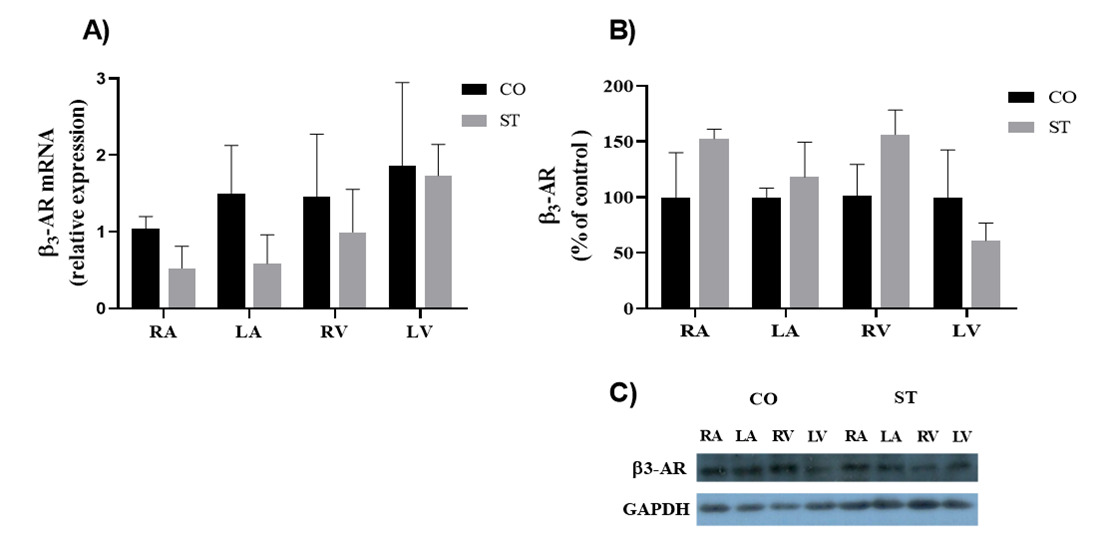
In agreement with the functional approach, Figure 3 shows that the β3-AR expression, as accessed in the four cardiac chambers, was not significantly altered by stress both at the mRNA and protein levels.
Nitric oxide synthase isoforms expression is reduced by stress
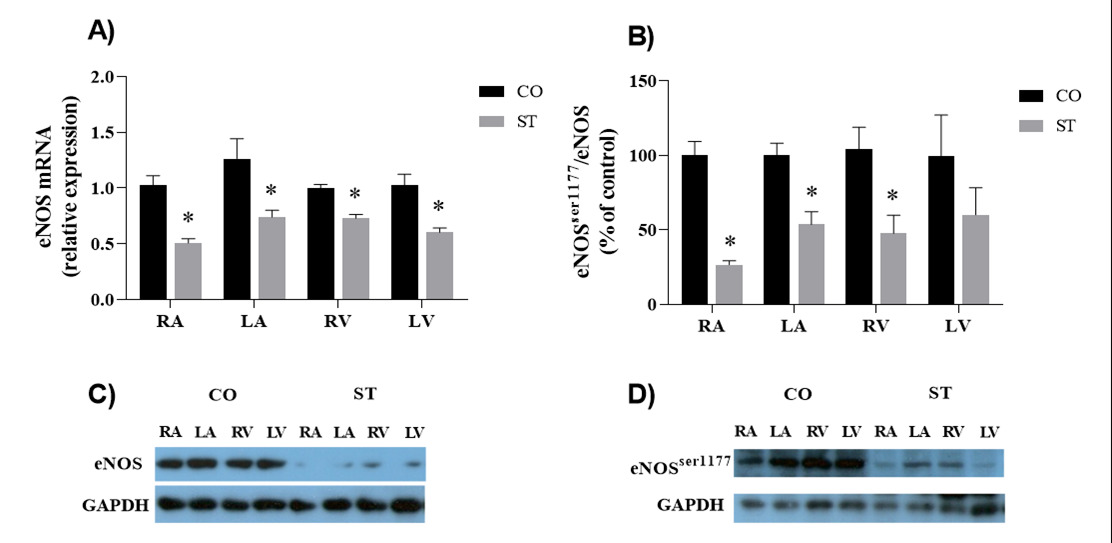
As above mentioned, the negative inotropic effect of β3-AR agonists is mediated by the agonist-receptor coupling to Gi/o protein, activation of NOS and consequent NO production [14,19]. Three isoforms of NOS are expressed in the heart [14,15]. The mRNA and protein expressions of total eNOS were similar in both ventricles and atria and were reduced in the heart of rats submitted to stress (Figure 4, panels A and C). eNOS activation depends on its phosphorylation. Data expressed in Figure 4 show that eNOSser1177 was reduced in ventricles and atria of rats submitted to stress (Figure 4, panels B and D). As a consequence, eNOSser1177/ total eNOS ratio was lower in the heart of stressed rats.
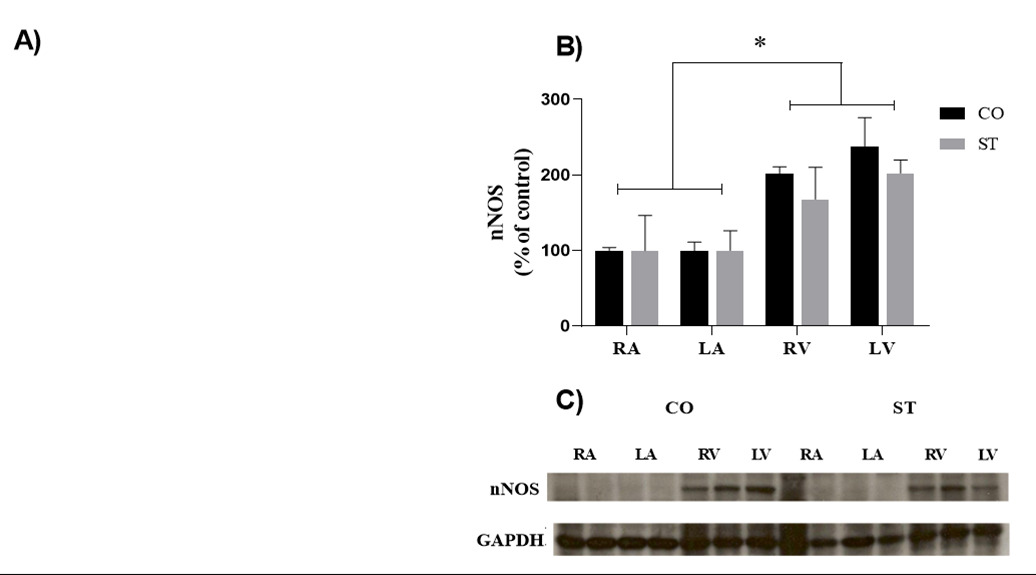
The mRNA expression of nNOS was similar in the cardiac chambers; however, nNOS protein expression was higher in the right and left ventricles of both groups in comparison with atria. Moreover, nNOS expression was not altered by foot-shock stress (Figure 5).
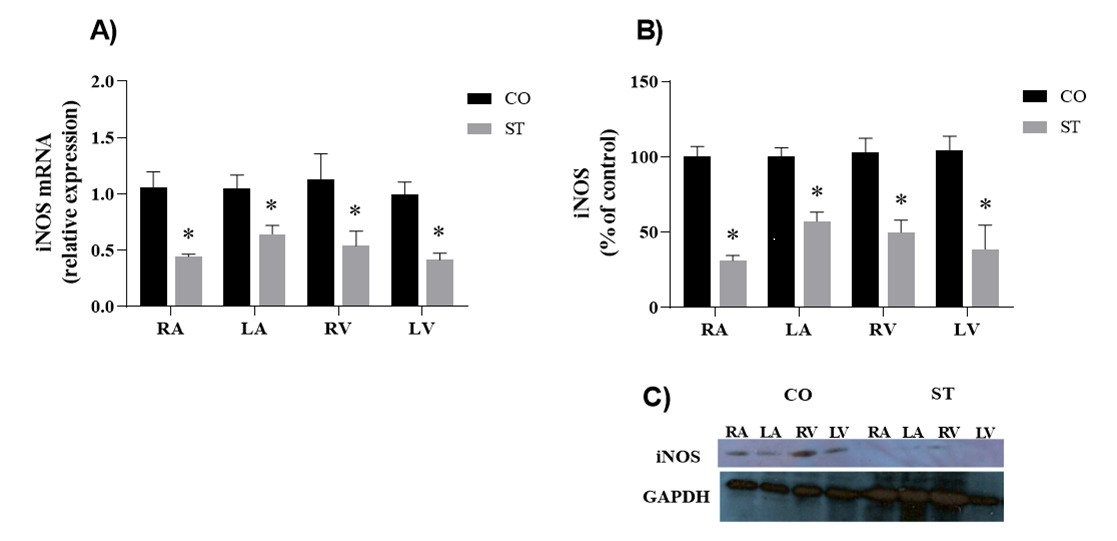
The inducible isoform of NOS, iNOS, had similar expression in all four heart chambers with consistent reduction of protein and gene expression in the heart of stressed rats compared to control (Figure 6).
Discussion
Data here presented have shown that β3-ARs were detected in both atria and ventricles of rats and that BRL 37, 344, in the range of nanomolar concentrations, had a negative inotropic effect of about 5-10% in the tension developed by the left atria. This effect was abolished by L-NAME. At the same concentration range, BRL 37, 344 had no effect in the beating rate of the isolated right atria.
Some other studies fail to show any effect of BRL 37, 344 on inotropism [17,28]. Despite that, it is now accepted that the cardiac β3-AR stimulation produces an inotropic effect in the myocardium, that is more pronounced in the ventricles and it is opposite to the one mediated by β1- and β2-AR [29]. This effect is due to the β3-AR activation of eNOS and nNOS, by means of Gi protein, thus increasing NO production; NO, in turn, activates a soluble form of guanylate cyclase leading to cGMP production followed by PKG activation that ultimately induces myocyte relaxation and causes negative inotropy, through the phosphorylation of troponin I, phospholamban, and L-type Ca2+ channels [29].
In the range of micromolar concentrations, BRL 37, 344 caused positive chronotropic and inotropic effects in right and left atria, respectively. These BRL 37, 344 effects were unaffected by L-NAME. Positive chronotropic effect of BRL 37, 344 in the right atria has been previously reported to be due to an increase in the intracellular level of cAMP as a consequence of BRL 37, 344 coupling to β2-AR- AC and stimulation of L-type Ca2+ channel-mediated current in isolated human atrial myocytes upon stimulation with BRL37344 [30].
Unlike the other two subtypes, β3-AR is expressed at a low level in the normal heart [4-6] and has a low tendency to desensitization because it lacks sites for phosphorylation by PKA and β-adrenoceptor kinase (βARK) [28,31]. Nevertheless, several reports showed changes in its expression at both protein and transcript level in pathophysiological conditions such as diabetes, heart failure, sepsis, and myocardial fibrosis [32-34]. However, although the stress protocol here used have modified the expression of β1- and β2-AR [26], it failed to alter the expression of β3-AR in any of the cardiac chambers or the sensitivity to BRL 37, 344.
As above mentioned, NOS is the downstream target of cardiac β3-AR-Gi/o protein [4-6]. Three isoforms of NOS are expressed in the healthy cardiomyocytes [5,6]. nNOS and eNOS generate and release NO in small quantities (< 100 nM) in the heart and are tightly controlled at post transcriptional level [8,35]. In contrast to cNOS that can be activated by calcium and calmodulin (CaM) in healthy states, iNOS can only be induced by inflammatory stimuli including immunostimulatory cytokines, bacterial products, or infection in different types of cells [7,36,37]. iNOS produces larger amounts of NO (> 1µM) and is primarily regulated at transcriptional level [8,35].
Where as eNOS protects the heart under pathological stress from hypertrophy and remodeling [30], iNOS-derived NO plays a cardio protective role via its antioxidant and vasodilator effects in normal physiological conditions [38]. eNOS deficient mice exhibited worse systolic and diastolic function and mortality after myocardial infarction when compared to wild type mice [19]. Therefore, maintaining eNOS expression and activity is beneficial by preventing the early stage of disease progression in the heart. It is also important to control iNOS activity since enhanced iNOS/NO production due to sympathetic nervous system activity might led to the formation of peroxynitrite, one byproduct of NO degradation, and its associated oxidative stress, which mediates the detrimental effects of iNOS/NO and have also been proved to induce necrosis and apoptosis in cardiomyocytes, being one of the important causes in myocardial damage [30,38], decreased myocardial function and survival. The expression of eNOSser1177 and iNOS was reduced in the heart of stressed rats. This will probably lead to less cardioprotective mechanisms and low systolic and diastolic function.
However, nNOS expression was unaltered by stress as shown by the present data. nNOS is expressed in the autonomic nervous system innervating the heart, aortic and pulmonary arteries, coronary artery, and the atrial and ventricular myocardium [39,40]. As such, nNOS is well placed to play essential roles in modifying sympathetic and parasympathetic tones, controlling heart rate, delivering essential nutrients through coronary arteries, and regulating myocardial contractility. In the myocardium, nNOS is predominantly localized in the sarcoplasmic reticulum (SR) [6] and is involved in the Ca2+ handling processes of cardiac excitation-contraction coupling [29,38]. Furthermore, nNOS in the SR translocates to the caveolae to protect the myocardium from Ca2+ overload and oxidative stress [41]. In addition, nNOS interacts with α-syntrophin through the scaffolding protein postsynaptic density-95 (PSD95) via the PSD-95/Discs large/ZO-1 homology domain (PDZ domain) and forms a multi-protein complex with the plasma membrane Ca2+ pump (PMCA) and voltage-gated Na+ channel (Nav1.5) [42,43]. In addition, nNOS binds to its PDZ-binding motif to direct nNOS to the sub cellular compartments, as is the case of nNOS in the nucleus [43], which regulates the transcription and activation of the elements required for oxidative phosphorylation and mitochondrial biogenesis [44].
The stress-induced contrasting tendency in protein expression of eNOS/iNOS and nNOS, that is the reduction in eNOS and iNOS proteins expression and activity whereas nNOS expression remained unaltered, plus the co-existence of eNOS, iNOS, and nNOS in the myocardium and their translocation, transcription, and post-translational modifications underlie the complex scenario of NO in the heart [39,45]. This contrasting tendency between eNOS and nNOS was more pronounced in the failing myocardium and in the hypertensive heart; that is, eNOS protein expression is reduced significantly, whereas nNOS protein expression and activity are increased [39,37]. Intriguingly, both eNOS and nNOS affect intracellular Ca2+ handling in the myocytes, and eNOS mediates spontaneous Ca2+ sparks and enhanced Ca2+ transients in cardiac myocytes in response to increased preload (mechanical stretch) [37,46]. Conversely, nNOS (but not eNOS) mediates the after load-induced spontaneous Ca2+ sparks [37].
Conclusion
So, the data presented here showed that the cardiac β3-AR expression levels were not altered by the stress protocol here used and that although there was a decrease in the expression and activity of eNOS as well as in the expression of iNOS, the method here used did not detect any alteration in the atria sensitivity to the β3-AR agonist BRL 37, 344. If the stress-induced reduction on eNOS and iNOS expressions would impair the cardioprotective mechanisms and the systolic and diastolic functions remains to be investigated. The nNOS sustained expression adds complexity to this scenario.
Acknowledgement
The authors wish to thank Flavia Pidone for the technical assistance and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil; São Paulo Research Foundation (FAPESP 2014/00314-8; 2016/20777-8; 2016/20784-4); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq312946/2014-7; 424114/2016-0) that supported this work.
Conflict of Interest Disclosure
The authors report no conflicts of interest.
References
- Juberg EN, Minneman KP, Abel PW (1985) ß1 and ß2-adrenoceptor binding and functional response in right and left atria of rat heart. Naunyn Schmiedebergs Arch Pharmacol 330: 193-202.
- Kaumann AJ, Molenaar P (1997) Modulation of human cardiac function through 4 ß-adrenoceptor populations. Naunyn Schmiedebergs Arch Pharmacol 355: 667-681.
- Woo AYH, Xiao R (2012) ß-Adrenergic receptor subtype signaling in heart: from bench to bedside. Acta Pharmacol Sin 33: 335-341.
- Emorine LJ, Marullo S, Briend-Sutren MM, et al. (1989) Molecular characterization of the human beta 3-adrenergic receptor. Science 245: 1118-1121.
- Gauthier C, Tavernier G, Charpentier F, et al. (1996) Functional beta3-adrenoceptor in the human heart. J Clin Invest 98: 556-562.
- Gauthier C, Langin D, Balligand JL (2000) Beta3-adrenoceptors in the cardiovascular system. Trends Pharmacol. Science 21: 426-431.
- Gauthier C, Rosec B, Manoury B, et al. (2011) Beta-3 adrenoceptors as new therapeutic targets for cardiovascular pathologies. Curr Heart Fail Rep 8: 184-192.
- Skeberdis VA, Gendviliene V, Zablockaite D, et al. (2008) Beta3-adrenergic receptor activation increases human atrial tissue contractility and stimulates the L-type Ca2+ current. J Clin Invest 118: 3219-3227.
- Rozec B, Erfanian M, Laurent K, et al. (2009) Nebivolol, a vasodilating selective beta(1)-blocker, is a beta(3)-adrenoceptor agonist in non-failing transplanted human heart. Journal of American College of Cardiology 53: 1532-1538.
- Triposkiadis F, KarayannisG, Giamouzis G, et al. (2009) The Sympathetic Nervous System in Heart Failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 54: 1747-1762.
- Brodde O (1991) Beta 1-and beta 2-adrenoceptors in the human heart: Properties, function, alterations in chronic heart failure. Pharmacol Rev 43: 203-242.
- Brodde OE, Michel MC (1999) Adrenergic and muscarinic receptors in the human heart. Pharmacological Reviews 51: 651-690.
- Spadari RC, Cavadas C, De Carvalho AETS, et al. (2018) Role of beta-adrenergic receptors and sirtuin signaling in the heart during aging, heart failure, and adaptation to stress. Cell Mol Neurobiol 38: 109-120.
- Gauthier C, Leblais V, Kobzik L, et al. (1998) The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest 102: 1377-1384.
- Zhen J, Lu H, Wang XQ, et al. (2008) Upregulation of endothelial and inducible nitric oxide synthase expression by reactive oxygen species. Am J Hypertens 21: 28-34.
- Irie T, Sips PY, Kai S, et al. (2015) S-Nitrosylation of Calcium-Handling Proteins in Cardiac Adrenergic Signaling and Hypertrophy. Circ Res 117: 793-803.
- Arioglu-Inan E, Kayki-Mutlu G, Michel MC (2019) Cardiac ß3-adrenoceptors - A role in human pathophysiology? Br J Pharmacol 176: 2482-2495.
- Birenbaum A, Tesse A, Loyer X, et al. (2008) Involvement of ß3-adrenoceptor in altered ß-adrenergic response in senescent heart: role of nitric oxide synthase 1–derived nitric oxide. Anesthesiology 109: 1045-1053.
- Balligand JL (2016) Cardiac salvage by tweaking with beta-3-adrenergic receptors. Cardiovasc Res 111: 128-133.
- Rehsia NS, Dhalla NS (2010) Mechanisms of the beneficial effects of beta-adrenoceptor antagonists in congestive heart failure. Exp Clin Cardiol 15: e86-e95.
- Lymperopoulos A, Rengo G, Koch WJ (2013) Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ Res 113: 739-753.
- Vielma AZ, Léon L, Fernandéz IC, et al. (2016) Nitric Oxide Synthase I modulates basal and ß-adrenergic-stimulated contractility by rapid and reversible redox-dependent S-nitrosylation of the heart. PLoS One 11: e0160813.
- Irie T, Sips PY, Kai S, et al. (2015) S-Nitrosylation of calcium-handling proteins in cardiac adrenergic signaling and hypertrophy. Circ Res 117: 793-803.
- Tamargo J, Caballero R, Gomez R, et al. (2010) Cardiac electrophysiological effects of nitric oxide. Cardiovasc Res 87: 593-600.
- Hoshida S, Yamashita N, Otsu K, et al. (2002) The importance of manganese superoxide dismutase in delayed preconditioning: involvement of reactive oxygen species and cytokines. Cardiovasc Res 55: 495-505.
- Moura ALD, Hyslop S, Grassi-Kassisse DM, et al. (2017) Functional ß2-adrenoceptors in rat left atria: effect of foot-shock stress. Can J Physiol Pharmacol 95: 999-1008.
- Santos IN, Spadari-Bratfisch RC (2001) Chronotropic response to (+/-)- CGP12177 in right atria of stressed rats. Can J Physiol Pharmacol 79: 393-399.
- Treinys R, Zablockaite D, Gendviliene V, et al. (2014) ß3-adrenergic regulationsof L-type Ca2+current and force of contraction in human ventricle. J Membr Biol 47: 309-318.
- Schena G, Caplan MJ (2019) Everything you always wanted to know about ß3-AR* (*but were afraid to ask). Cells 8: 357-381.
- Sterin-Borda L, Bernabeo G, Ganzinelli S, et al. (2006) Role of nitric oxide/cyclic GMP and cyclic AMP in ß3 adrenoceptor-chronotropic response. J Mol Cell Cardiol 40: 580-588.
- Oseke K, Angers S, Bouvier M, et al. (2019) Agonist-induced desensitization of ß3-adrenoceptors: Where, when and how? Br J Pharmacol 176: 2539-2558.
- Hermida N, Michel L, Esfahani H, et al. (2018) Cardiac myocyte 3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling. Eur Heart J 39: 888-898.
- Dincer UD, Bidasee KR, Guner S, et al. (2001) The effect of diabetes on expression of ß1-, ß2-, and ß3-adrenoreceptors in rat hearts. Diabetes 50: 455-461.
- Moniotte S, Kobzi L, Feron O, et al. (2001) Up regulation of ß3-adrenoceptorsand altered contractile response to inotropic amines in human failing myocardium. Circulation 103: 1649-1655.
- Murphy E, Kohr M, Sun J, et al. (2012) S-nitrosylation: A radical way to protect the heart. J Mol Cell Cardiol 52: 568-577.
- Hess DT, Matsumoto A, Kim SO, et al. (2005) Protein S-nitrosylation: Purview and parameters. Nat Rev Mol Cell Biol 6: 150-166.
- Hare JM (2004) Nitroso-redox balance in the cardiovascular system. N Engl J Med 351: 2112-21144.
- Yu XY, Ge L, Niu L, et al. (2018) The dual role of inducible Nitric Oxide Synthase in Myocardial Ischemia/Reperfusion Injury: Friend or Foe? Oxid Med Cell Longev 2018: 8364848.
- Barouch LA, Harrison RW, Skaf MW, et al. (2002) Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416: 337-339.
- Zhang YH, Jin CZ, Jang JH, et al. (2014) Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J Physiol 592: 3189-200.
- Zhang YH, Casadei B (2012) Sub-cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol 52: 341-350.
- Sun J, Picht E, Ginsburg KS, et al. (2006) Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+channel alpha 1 subunit and reduced ischemia/reperfusion injury. Circulation Research. 98: 403-411.
- Ueda K, Valdivia C, Medeiros-Domingo A, et al. (2008) Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A 105: 9355-9360.
- Aquilano K, Baldelli S, Ciriolo MR (2014) Nuclear recruitment of neuronal nitric- oxide synthase by a-syntrophin is crucial for the induction of mitochondrial biogenesis. J Biol Chem 289: 365-378.
- Zhang YH (2016) Neuronal nitric oxide synthase in hypertension: An update. Clin Hypertens 22: 20
- Petroff MG, Kim SH, Pepe S, et al. (2001) Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+release in cardiomyocytes. Nat Cell Biol 3: 867-873.
Corresponding Author
Regina CeliaSpadari, Department of Biosciences, Laboratory of Stress Biology, Institute of Health and Society, Campus Baixada Santista, UNIFESP, Rua Silva Jardim 136, Santos São Paulo, Brazil, CEP 11015-020.
Copyright
© 2022 Caceres V. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.