Trichorhinophalangeal Syndrome Type 1: Unusual Case Report and Review of Literature
Abstract
Trichorhinophalangeal syndrome type 1 [TRPS1; (OMIM #190350)] is a rare autosomal dominant genetic disorder on chromosome 8q24. The carrier of the syndrome may present skeletal, craniofacial, and oral manifestations. The main characteristics that the individual may present are scarce hair or total alopecia, thin upper lip, long erased nasolabial filter, bulbous nose and low set ears. In the oral cavity, there may be changes such as supernumerary teeth and hypodontia, size changes, including macrodontia and microdontia, and teeth in malocclusion. This study aims to review the literature on the main characteristics of TRPS1 and present a rare case with systemic and especially intraoral phenotypic characteristics of a pediatric patient with trichorhinophalangeal syndrome type 1 different from those already reported in literature. Twenty-seven articles dealing exclusively with TRPS1 were reviewed and its main systemic and intraoral characteristics were described. TRPS1 is rare, and the knowledge of new phenotypic characteristics is important to facilitate the diagnosis.
Keywords
Trichorhinophalangeal Syndrome Type 1, Oral Manifestations, Mutation
Introduction
Trichorhinophalangeal syndrome type 1 [TRPS1; (OMIM #190350)] is characterized by being a rare autosomal dominant alteration due to chromosome 8q24 mutation or deletion, in the TRPS1 gene [1]. It is suggested that this gene may be involved with the regulatory activity of bone mineralization, besides the maturation and apoptosis of chondrocytes [2]. Andreas Giedion and Leonard O Langer Jr were the first researchers to describe the syndrome in 1966 [3]. Its prevalence is 1:1,000,000, being more common in females (7:1) [4].
Because it presents a broad description of abnormalities, TRPS is classified into three subtypes according to the level of gene mutation. In addition to the typical phenotype of the syndrome, patients with subtype 1 may present short stature and bone malformations. In subtype 2, depending on the degree of deletion of the TRPS1, the individual may have mental retardation and bone exostosis. TRPS subtype 3 is characterized as a more intense manifestation of subtype 1, and the individual may have severe brachydactyly and delayed growth [5].
The carrier of the syndrome may present multisystemic alterations such as multiple osteochondromas, conical-shaped bone epiphyses, short stature and a greater propensity to developing respiratory and urinary infections [6]. A typical facial phenotype includes total alopecia or the presence of scarce hair, bulbous nose shaped like a pear, thin upper lip, long and erased nasolabial filter, and low ear implantation [7].
The TRPS1 mutation may be related to problems during odontogenesis, and this gene acts on the formation of dental follicles [8]. Thus, changes in the number of teeth, such as supernumerary and hypodontia, size changes, including microdontia or macrodontia, and teeth in malocclusion may occur [9]. The differential diagnoses are ectodermal dysplasia, Larsen's syndrome and digital orofacial syndrome that present clinical characteristics similar to TRPS1, such as the typical facial phenotype with prominent forehead, bulbous nose, long nasolabial filter, low ear implantation, the presence of supernumerary teeth and skeletal abnormalities [10,11].
Considering these aspects and due to the rarity of the syndrome, this study aims to describe a case report presenting new phenotypic characteristics of TRPS1 with emphasis on the oral cavity and review the literature on phenotypic characteristics of the syndrome already known.
Methods
This review was performed to describe the phenotypic characteristics published to date and to highlight characteristics not yet reported in a patient affected by TRPS1.
Search strategies
An electronic search on Pubmed (MEDLINE), Scopus, Google Scholar and The Cochrane Library was carried out by two independent reviewers until December 2018, when we searched the following terms: 'trichorhinophalangeal syndrome type 1 and orofacial features' [title/abstract] focusing on the oral and systemic characteristics.
Inclusion and exclusion criteria
Sixty-six studies were originally found through the literature search strategy, and six additional citations were identified from the references of potentially recovered studies. Forty-five papers were excluded for various reasons (duplication in the databases, absence of relationship with the proposed theme, unavailability of the full texts, or because they are comments or expanded abstracts). All other studies were selected to evaluate the characteristics of patients with TRPS1.
The inclusion criteria used to identify relevant articles were: (i) Cases that directly addressed the oral manifestations of TRPS1; (ii) Cases that generally address only patients affected by TRPS1, and (iii) Genetically confirmed diagnosis of TRPS1. All studies that included individuals with alterations or syndromes other than TRPS1 were excluded. Thus, 27 studies were selected for their complete reading.
Prospective or retrospective cohort studies, case series and case reports describing clinical and/or laboratorial characteristics of patients with dental anomalies in TRPS1 were included. The titles and abstracts of all reports in the electronic searches were individually read by two investigators. The studies that fulfilled the inclusion criteria and those that did not present sufficient information in the title or in the abstract were fully assessed. A crosscheck assessment of the articles was performed by another author to guarantee that the proper studies were selected according to the inclusion and exclusion criteria.
Data extraction
We independently collected study data through specific extraction forms by using Microsoft Excel software®. For the studies selected, the following information was extracted: Year and country of publication, type of article, number of cases, sex of the patients, systemic manifestations and oral alterations described by the authors.
Analysis
All the clinical information of the patients and diagnostic criteria were evaluated for descriptive cross-referencing of the data, considering the information given by the authors, where the main oral and systemic alterations were observed in patients affected by TRPS1.
Case Report
A 10-year-old female Caucasian patient was referred to the Center for Rehabilitation of Craniofacial Anomalies, Minas Gerais, Brazil. The patient has been assisted since two months of age, when her parents sought the clinic due to the absence of nose cartilage. Her mother reported not noticing abnormalities during the pregnancy and denied consanguinity with her husband; the delivery occurred normally. At birth, it was possible to notice the presence of a unilateral left cleft lip and also the dehiscence of cartilage in the nasal region, which caused respiratory difficulties to the patient. Thus, rhinoplasty and septoplasty were performed due to respiratory and aesthetic problems. The patient also underwent surgery to remove the adenoid gland, in addition to surgical procedure to lift the eyelids. The first diagnostic hypothesis was craniofacial fibrous dysplasia due to the severe craniofacial alterations observed.
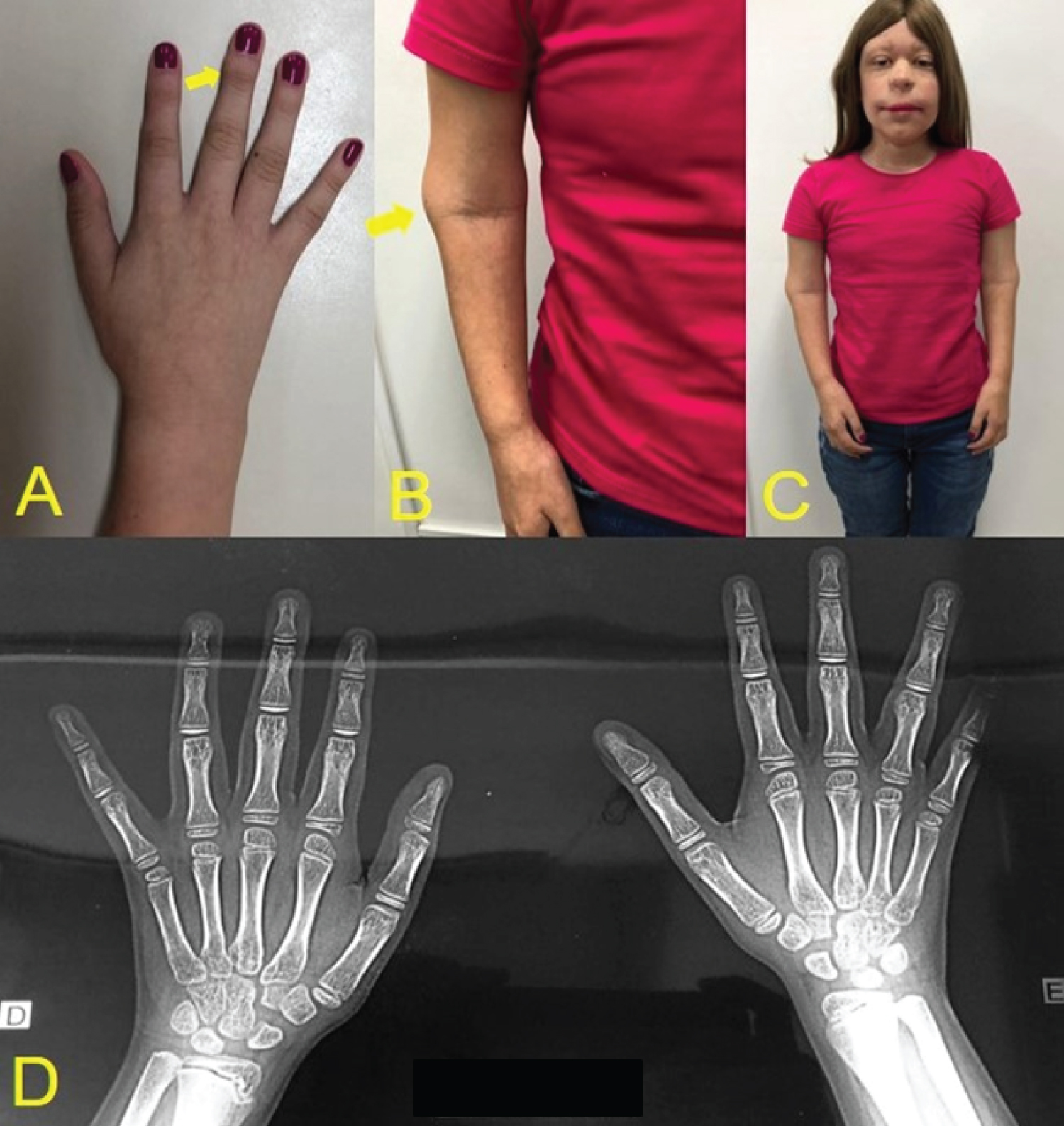
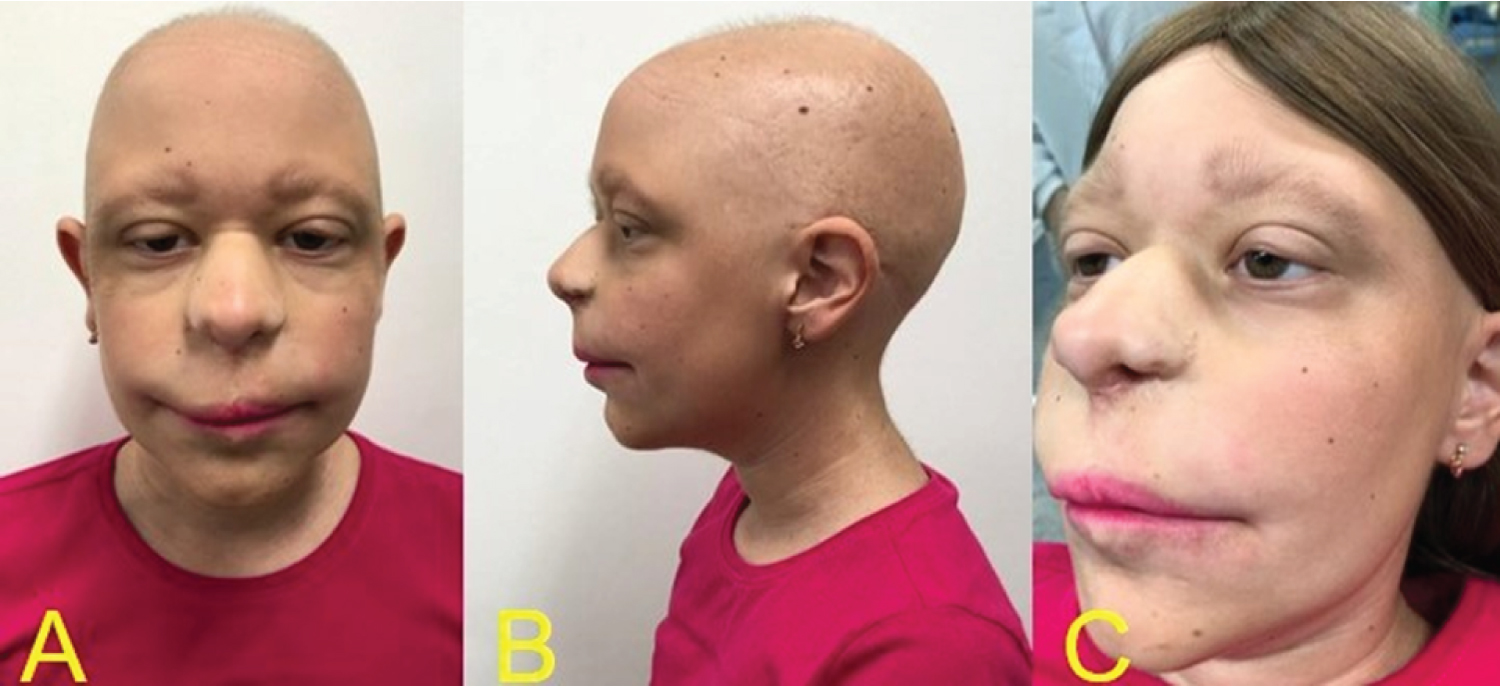
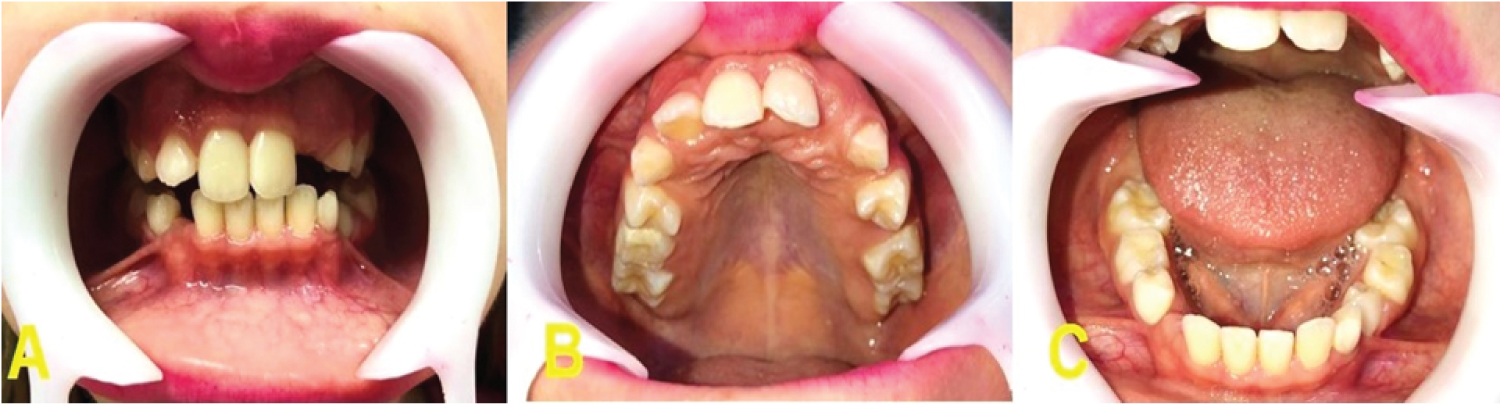
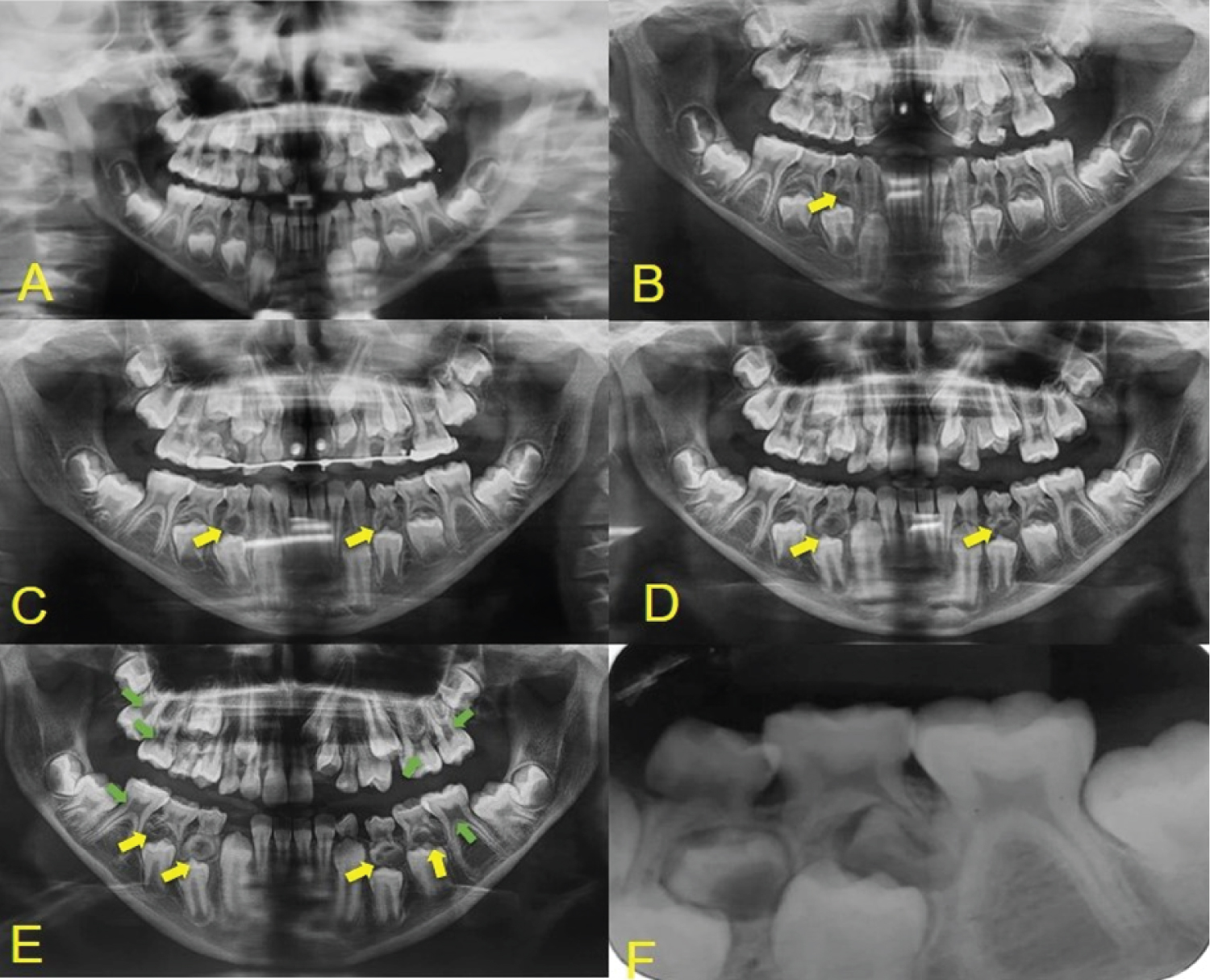
Throughout the patient's follow-up, different characteristics from other syndromes were noticed, which did not correspond to the previous diagnoses, and after being referred to a genetic medical center, the diagnosis of non-sense mutation in TRPS1 gene in chromosome 8q24, was confirmed through the karyotyping test. Among the systemic alterations, the patient presented short stature, arthritis in the arms making them impossible to move, cone-shaped bone epiphyses of the hands, and changes in the hip (Figure 1). The patient presents intellectual development compatible with her age. In the facial manifestations it is possible to observe generalized alopecia with sparse hair in the region of the scalp and eyebrows, madarosis, eyelid ptosis, low implantation of the ears, and nose with bulbous shape (Figure 2). Changes in the oral cavity include the development of supernumerary teeth, hyperplastic gums (Figure 3) and type 2 taurodontism in the lower and upper molars. The patient has already undergone exodontia of seven supernumerary teeth and gingivectomy in the region of the right lateral incisor. Based on the most recent panoramic radiographs, four more supernumerary tooth germs were formed in the posterior region of the mandible and maxilla (Figure 4).
Results
Twenty-seven papers that directly and exclusively addressed the TRPS1 were found (Table 1), comprising reports and case series, prospective and retrospectives studies, all with confirmed genetic studies for diagnostic syndrome. In addition, a case with rare phenotypic manifestations of a pediatric patient diagnosed with TRPS1 has been inserted. Besides the usual manifestations of the syndrome, such as short stature, joint alterations, and conical bone epiphyses of the hands, new manifestations including the presence of oral clefts, lack of cartilage of the nose, what may be a proposal to increasing phenotypic spectrum. Besides the usual manifestations of the syndrome, new manifestations including the presence of oral cleft, lack of cartilage of the nose and telangiectasia on the epithelium of the skin were also found, what may be a proposal to increasing the phenotypic spectrum of the syndrome.
A descriptive (Table 1) was made with of all 27 selected papers, dividing the manifestations into systemic, craniofacial, and oral, thus comparing the patients characteristics that were compatible with the syndrome and adding other characteristics that were not described in the papers analyzed.
Discussion
TRPS1 is an autosomal dominant condition caused by the mutation or deletion of chromosome 8q24 present in the TRPS1 [1]. Due to its rarity and its different degrees of expression in patients, it is clinically and radiographically unfeasible to diagnose it, and genetic testing is essential [12,13]. Clinically, it is possible to find alterations such as short stature, brachydactyly, syndactyly, short hands and feet, and abnormal nails [14-22]. Among the craniofacial alterations, scarce hair with slow growth, bulbous nose, long and erased nasolabial filter, thin upper lip and low implantation of the ears stand out [21,23-29]. Teeth in malocclusion and supernumerary are the most common oral manifestations [30,31]. The most common radiographic finding is cone-shaped bone epiphyses of the hands, besides the occurrence of hip and joint abnormalities [2,32]. Bone exostoses are uncommon in TRPS1 and more common in type 2, but it may occur in isolation [33].
Analyzing the cases, it was observed that there is female predilection and no ethnic group predilection. There are some clinical signs that are manifested in their carriers in a homogeneous way [34,35]. Bone epiphyses in the conical shape of the middle phalanges of the hands occur in most cases, being the main sign of the syndrome. Nasseli, et al. [16], Seitz, et al. [18] and Chen, et al. [19] report that generally the fingers are also affected by other deformities such as clinodactyly, brachydactyly, syndactyly, but none of these changes were found in our patient.
The low stature of our patient (Figure 1) also occurs in most patients as shown by Flores-Cuevas, et al. [21], Sohn, et al. [1] and Nan, et al. [25]. The typical facial phenotype of the syndrome is present in all the clinical cases analyzed and is in line with the facial phenotype of the case presented [2,13-37]. Although mental retardation and bone exostosis are typical characteristics of type 2 syndrome, they were present in type 1 syndrome in the cases of Brodwall, et al. [20] and Kunotai, et al. [33], respectively. These characteristics may be expressed according to the size of the deletion on chromosome 8q [7].
The most striking manifestation of the syndrome in the oral cavity are claimed to be the development of supernumerary teeth. The case of Kunotai, et al. [33] showed that one of the patients underwent exodontia of 17 supernumerary teeth, which is the most significant case analyzed in this study. Our patient has supernumerary teeth, seven of which have already been removed (Figure 4). It is highlighted that the radiograph performed in 2019 shows the development of four new supernumerary tooth germs in the region of the lower molars and the presence of taurodontism in the lower and upper first molars contrasting with the panoramic radiograph taken in 2016 (Figure 4E). This condition was not reported in any of the cases observed in this study, since generally after exodontia there is no development of new supernumerary teeth. Thus, the manifestations in the oral cavity should be taken into consideration when establishing the diagnosis of TRPS1.
Regarding the presence of unilateral left lip cleft and absence nasal cartilage in our patient, we noted that only Maas, et al. [32] report that there was underdevelopment of the nasal malar region, but not the complete absence of cartilage. Due to the absence of cases in literature relating the presence of lip cleft with the TRPS1, this diagnostic hypothesis of the syndrome was not considered at birth. Thus, the diagnosis was defined based on the G-band karyotype test, which included translocation of chromosome 8q22-24.
With the diagnosis defined, it was possible to observe that the patient showed other signs that are not common or have not been described in literature, what may expand the phenotypic spectrum of TRPS1. Continuous development of supernumerary teeth in the mandibular region was also observed, with taurodontism of the upper and lower molars and hyperplastic gums. Comparing the findings in our case to those reported in the studies observed in this paper, it is possible to associate new manifestations with TRPS1, especially in the head and oral tract. Systematically, the telangiectatic skin and elongated fingers stand out instead of shortening, as reported in most cases [2,30-35]. Other manifestations such as short stature, joint and hip problems coincide with the reports of Jeon, et al. [29] and Merjaneh, et al. [31].
The presence of cleft lip had not been correlated as part of the spectrum of patients with TRPS1, as well as the dehiscence of cartilage in the nose and palpebral eyelid ptosis. Some other signs potentially associated with the syndrome but never described in literature such as the hyperplastic gingiva, the continuous development of supernumerary teeth and the occurrence of taurodontism in the lower and upper molars were also observed in our patient. The knowledge of new phenotypic characteristics is important to facilitate early diagnosis, what may offer a better attention and survival to the patient.
Acknowledgments
The Minas Gerais State Research Foundation -Fapemig, Brazil and National Council for Scientific and Technological Development - CNPq, Brazil.
Conflict of Interest
None declared.
Ethical Approval
Approval for this retrospective study was granted by the research ethics committee of the Jose do RosárioVellano University (Registration number: 3.392.996).
Patient Consent
The dissemination of images and information on the identification of the patient by the responsible persons was authorized.
References
- George S, Pulimood S, Korah I (1998) Trichorhinophalangeal syndrome type I. J Eur Acad Dermatol Venereol 11: 66-68.
- De Barros GM, Kakehasi AM (2016) Skeletal abnormalities of tricho-rhino-phalangeal syndrome type I. Rev Bras Reumatol Engl Ed 56: 86-89.
- Giedion A (1966) Das tricho-rhino-phalangealesyndrome. Helv Paediatr Acta 21: 475-482.
- Dias C, Isidoro L, Santos M, et al. (2013) Trichorhinophalangeal syndrome type I: A patient with two novel and different mutations in the TRPS1 gene. Case Rep Genet 2013: 748057.
- Giedion A (1998) Phalangeal cone-shaped epiphyses of the hand: their natural history, diagnostic sensitivity, and specificity in cartilage hair hypoplasia and the trichorhinophalangeal syndromes I and II. Pediatr Radiol 28: 751-758.
- Michalek P, Doherty JT, Vesela MM (2009) Anesthetic management of a child with Langer-Giedion (TRPS II) syndrome. J Anesth 23: 456-459.
- Pilch J, Plomp A, Poke G, et al. (2015) Phenotype and genotype in 103 patients with tricho-rhino-phalangealsyndrome. Eur J Med Genet 58: 279-292.
- Goss M, Socorro M, Monier D, et al. (2019) Trps1 transcription factor regulates mineralization of dental tissues and proliferation of tooth organ cells. Mol Genet Metab 126: 504-512.
- Kantaputra P,Miletich I, Lüdecke HJ, et al. (2008) Tricho-rhino-phalangeal syndrome with super numerary teeth. J Dent Res 87: 1027-1031.
- Ferreira CS, Ferreira RAMH, Fernandes MLMF, et al. (2012) Displasia Ectodérmica: Relato de caso. Arquivos em Odontologia 48: 47-52.
- Piccione M, Niceta M, Antona V, et al. (2009) Identification of two new mutations in TRPS 1 gene leading to the tricho-rhino-phalangeal syndrome type I and III. Am J Med Genet A 149A: 1837-1841.
- Bennett CG, Hill CJ, Frias JL (1981) Facial and oral findings in trichorhinophalangeal syndrome type 1 (characteristics of TRPS 1). Pediatr Dent 3: 348-352.
- Noltorp S, Kristoffersson UL, Mandahl N, et al. (1986) Trichorhinophalangeal syndrome type I: Symptoms and signs, radiology, and genetics. Ann Rheum Dis 45: 31-36.
- Vora NS, Shah S, Dave JN, et al. (1995) Trichorhinophalangeal syndrome type 1. Indian J Dermatol Venereol Leprol 61: 111-112.
- Machuca G, Martínez F, Machuca C, et al. (1997) Craniofacial and oral manifestations of trichorhinophalangeal syndrome type I (Giedion's syndrome): a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 84: 35-39.
- Naselli A, Vignolo M, Di Battista E, et al. (1998) Trichorhinophalangeal syndrome type I in monozygotic twins discordant for hip pathology. Report on the morphological evolution of cone-shaped epiphyses and the unusual pattern of skeletal maturation. Pediatr Radiol 28: 851-855.
- George R, Najmuddin F, Rai R, et al. (2014) Langer-giedion syndrome: A distinct phenotype. Iran J Pediatr 24: 656-657.
- Seitz CS, Lüdecke HJ, Wagner N, et al. (2001) Trichorhinophalangeal syndrome type I: clinical and molecular characterization of 3 members of a family and 1 sporadic case. Arch Dermatol 137: 1437-1442.
- Chen LH, Ning CC, Chao SC (2010) Mutations in TRPS1 gene in trichorhinophalangeal syndrome type I in Asian patients. Br J Dermatol 163: 416-419.
- Brodwall KM, Júlíusson PB, Bjerknes R, et al. (2011) Trichorhinophalangeal syndrome--clinical presentation and genetics. Tidsskr Nor Laegeforen 131: 1420-1423.
- Flores-Cuevas A, Mutchinick O, Morales-Suárez JJ, et al. (2012) Identification of two novel mutations in TRPS1 gene in families with tricho-rhino-phalangeal type I syndrome. J Investig Med 60: 823-826.
- Durmus O, Cakar E, Ata E, et al. (2014) Cone-shapedepiphysis. Am J Phys Med Rehabil 93: 92-93.
- Sohn YB, Ki CS, Park SW, et al. (2012) Clinical, biochemical, and genetic analysis of two korean patients with trichorhinophalangeal syndrome type I and growth hormone deficiency. Ann Clin Lab Sci 42: 307-312.
- Candamourty R, Venkatachalam S, Karthikeyan B, et al. (2012) Trichorhinophalangeal syndrome type 1: A case report with literature review. J Nat SciBiol Med 3: 209-211.
- Nan X, Dai S, Li CT, et al. (2013) Novel deletion mutation of TRPS1 gene in a Chinese patient of trichorhinophalangeal syndrome type I. Gene 523: 88-91.
- Ghoneima A, Sachdeva K, Hartsfield J, et al. (2013) The use of cone beam computed tomography for the assessment of trichorhinophalangeal syndrome, type I - a case report. J Orthod 40: 47-52.
- Corsini T, Cuvillon P, Forgeot A, et al. (2013) Single-dose intraincisional levobupivacaine infiltration in caesarean postoperative analgesia in TRPS1: A placebo-controlled double-blind randomized trial. Ann Fr Anesth Reanim 32: 25-30.
- Hufeland M, Rahner N, Krauspe R (2015) Trichorhinophalangeal syndrome type I: A novel mutation and Perthes-like changes of the hip in a family with 4 cases over 3 generations. J Pediatr Orthop 35: e1-e5.
- Jeon J, Kim JH, Oh CH (2014) Trichorhinophalangeal syndrome type I- clinical, microscopic, and molecular features. Indian J Dermatol Venereol Leprol 80: 54-57.
- Fujisawa T, Fukao T, Shimomura Y, et al. (2014) A novel TRPS1 mutation in a family with tricho-rhino-phalangeal syndrome type 1. J Dermatol 41: 514-517.
- Merjaneh L, Parks JS, Muir AB, et al. (2014) A novel TRPS1 gene mutation causing trichorhinophalangeal syndrome with growth hormone responsive short stature: a case report and review of the literature. Int J Pediatr Endocrinol 2014: 16-21.
- Maas S, Shaw A, Bikker H, et al. (2015) Trichorhinophalangeal syndrome. J EurAcad Dermatol Venereol 24: 358-369.
- Kunotai W, Ananpornruedee P, Lubinsky M, et al. (2017) Making extra teeth: Lessons from a TRPS1 mutation. Am J MedGenet A 173: 99-107.
- Saito R, Yamaguchi T, Ohmori S, et al. (2018) A novel base pair deletion in the TRPS1 gene in a Japanese patient with trichorhinophalangeal syndrome. Eur J Dermatol 28: 107-108.
- Trippella G, Lionetti P, Naldini S, et al. (2018) An early diagnosis of trichorhinophalangeal syndrome type 1: A case report and a review of literature. Ital J Pediatr 44: 138-142.
- Su W, Shi X, Lin M, et al. (2018) Non-ossifying fibroma with a pathologic fracture in a 12-year-old girl with tricho-rhino-phalangeal syndrome: A case report. BMC Med Genet 19: 211-217.
- Nicolescu CR, Kasongo L, Rausin L (2018) Dysmorphic short stature: Radiological diagnosis of trichorhinophalangeal syndrome. Case Rep Pediatr 2018: 90-96.
Corresponding Author
Rodrigo Soares de Andrade, Oral Pathology, UNIPAM, Campus III, 2° Piso, Av. Marabá, Alto dos Caiçaras. Patos de Minas - MG, 38700-207, Brazil, Tel: 55-19-999665488
Copyright
© 2021 Andrade RS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.