Langerhans Cell Histiocytosis-A Case Report
Abstract
Langerhans Cell Histiocytosis (LCH) is among the group of diseases that was originally called Histiocytosis X. This syndrome has an unknown etiology and originates from an excessive proliferation of Langerhans cells. The histiocytes can cause the development of other pathological conditions. LCH has a predilection for men and children and is classified into the following three types: LCH with solitary lesion, multiple lesions and disseminated/with visceral involvement. A common feature of these variants is the presence of a lytic lesion that can be identified by imaging. This study aimed to report a clinical case of LCH with multiple lesions and wide systemic repercussions but with no significative oral alteration. The patient, a 31-year-old woman, leucoderma, presented with a more than 10-year history of pain in the lower limbs. It was requested imaging examinations to assess the presence of bone rarefactions in the femur and iliac. An incisional biopsy of the lesion revealed a well-differentiated fragment of bone tissue in the cortical and medullary regions, without histopathological changes. However, deposits of histiocytic cells with eosinophilic cytoplasm and multinucleated giant cells were present in some areas. A final histopathological diagnosis of LCH was established based on these findings. Since then, the patient's condition has been effectively controlled with bisphosphonate therapy. This case report might contribute to a better understanding of the pathogenesis of LCH and will help to expand the knowledge of health professionals, especially dental surgeons, about this condition.
Keywords
Histiocytosis X, Granuloma, Langerhans
Introduction
Langerhans Cell Histiocytosis (LCH) is characterized by clonal proliferation of histiocytes with granulomatous formation. The etiology of this syndrome is unknown. Histiocytes are dendritic, monomorphonuclear, antigen-presenting cells located in the epidermis, bone marrow, lymph nodes, and mucosa [1]. It is a rare disease with an incidence of 1 to 4 cases per 1,000,000 individuals, men's predilection, and occurs with a greater frequency in children and teenagers [1,2]. According to the latest WHO recommendations regarding the nomenclature, LCH is classified into three types: LCH with solitary lesion, multiple lesions and disseminated/with visceral involvement [3,4].
In the oral cavity, the first alterations of this clinical condition can be detected early as 10-20% of the maxillary bones are affected [2]. The most commonly affected site is the posterior region of the mandible, where severe periodontitis is observed because of increased dental mobility. Floating teeth can also be observed on panoramic radiographs because of moderate-to-intense resorption of alveolar processes [2,5,6]. In LCH with multiple lesions, the presence of a clinical triad consisting of exophthalmia, diabetes insipidus, and bone lesions is also observed in a minority of patients [7-9]. The initial inflammation can be diagnosed by a significant presence of histiocytes at the site, which is a histopathological manifestation that confirms the presence of the disease. Because this variant has disseminated characteristics, it is not restricted to the bone and might also affect oral mucosa and organs like liver, kidneys, and lymph nodes [2].
As an acute form of the disease, LCH with disseminated/with visceral involvement, occurs in children and young people. Mucosal and bone tissue are compromised, and diffuse histiocyte proliferation occurs throughout the body. In 1953, Lichtenstein, et al. [10] described that this syndrome could be fatal without any reponse to steroidal therapy and control of the clinical manifestation [1,5,9]. The third variant, LCH with solitary lesion, is restricted to the bone and may be unifocal or multifocal, and mild or severe. It generally does not present cutaneous, pulmonary, or pituitary manifestations. Moreover, its prognosis is more favorable than that of the other variants of LCH [2,4,5].
As it is a rare disease, periodontists should pay attention to the conduct of a patient with such lesions since the correct diagnosis is extremely important for the prevention of negative sequelae caused by periodontal dysfunction [6]. The diagnosis of this disease is confirmed by histopathological evaluation, consisting of an immunohistochemical examination of S100, Langerin and CD1-A proteins [8].
The treatment is determined according to the severity, size of the lesions, and systemic involvement. Corticosteroids, administered jointly, are the drugs of choice to treat this syndrome. According to literature, prednisolone and vincristine have been used concomitantly, with favorable prognosis [1]. In the oral cavity, the treatment is restricted to classic periodontal therapy with scaling and root planning, or a surgical approach. In both cases, periodic follow-up of the lesions by the periodontist is necessary [2,6]. Given the rarity of this dysfunction and the scarcity of studies published in literature emphasizing the oral cavity, this case report aims to describe an unusual case of HCL that affected a young adult patient with extensive systemic lesions, but with no involvement of the jaws.
Case Report
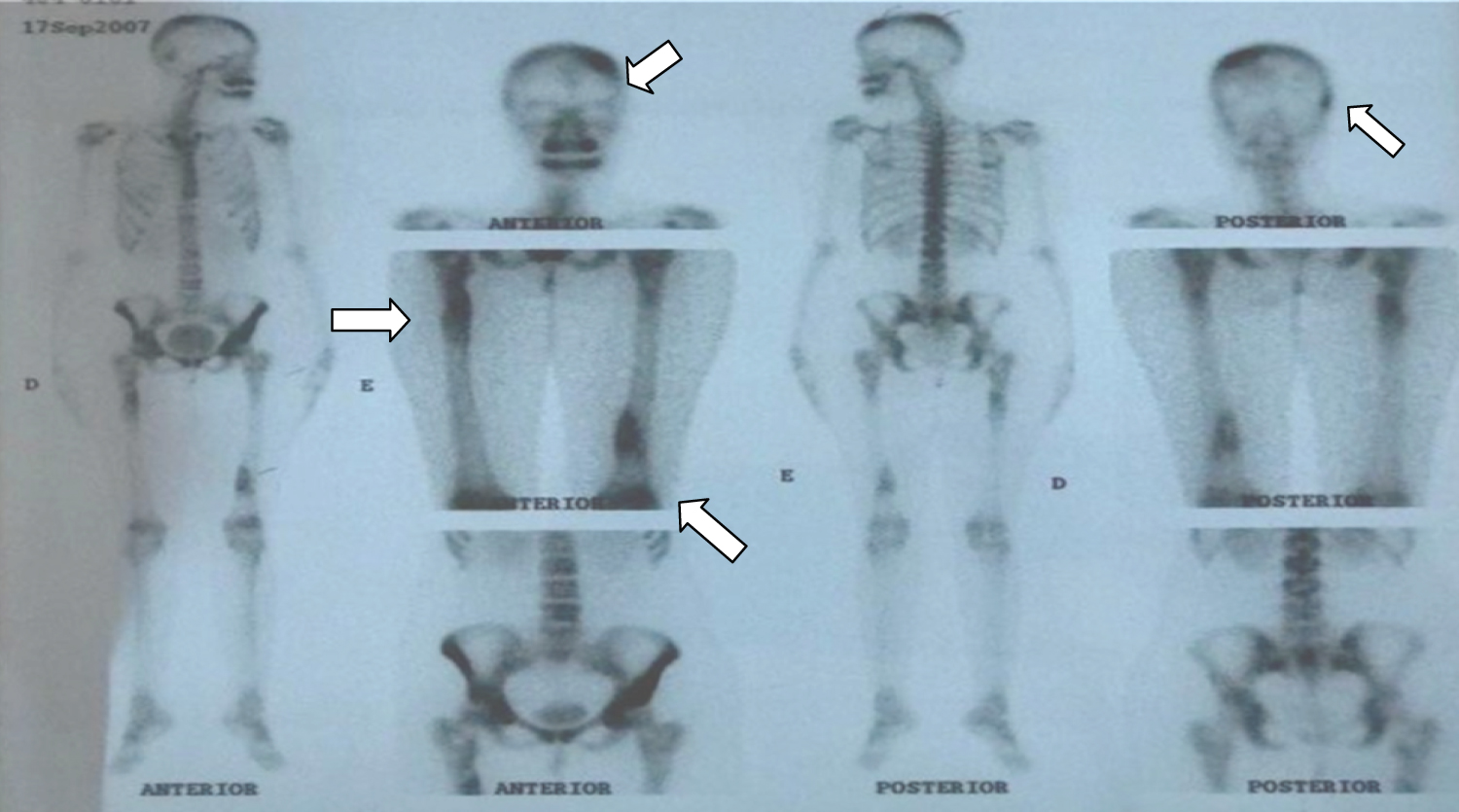
A 32 year-old woman, leucoderma, presented at an orthopedician's clinic with complaints of pain in the lower limbs, with increased pain in cold weather. At this time, the doctor did not request any complementary exams and said to the her family that it was a disease of late musculature growth. Two years later, in 2007, the pain worsened. The patient consulted a rheumatologist who examined her clinically and requested complementary tests, including the analysis of the hemosedimentation velocity serum values (VHS; 54 mm\1st hour), and positive C-reactive protein (PCR) (++). The anti-core factor (FAN) and rheumatoid factors, C3 and C4, showed normal values. On the same occasion, a radiographic examination of the lower limbs was also performed to better evaluate the clinical picture. Hashimoto syndrome, Paget's disease, and late muscle growth disease were considered as possible differential diagnoses. The images revealed bone rarefaction in the lower limbs. Additionally, on scintigraphy, osteogenic lesions that needed further clarification were found in the left parietal, right temporoparietal, left anteroinferior iliac spine, right femur proximal half, and the proximal and distal thirds of the left femur (Figure 1) arrows.
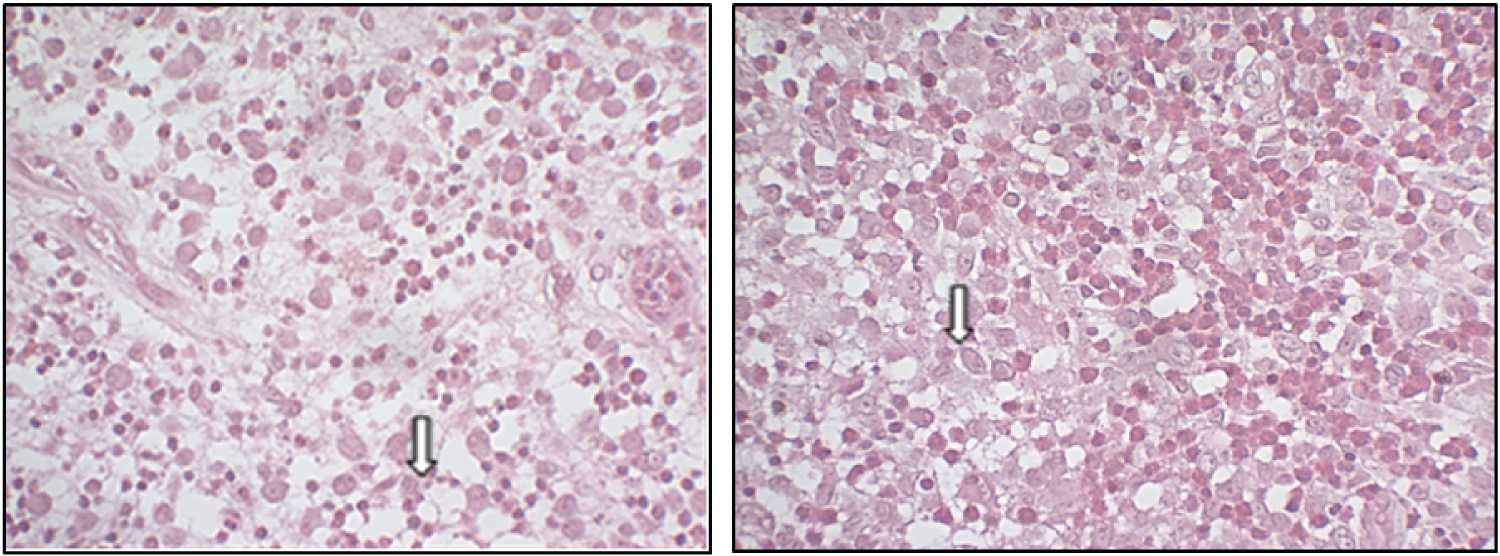
According to the clinical, imaging, and serological findings, the patient underwent a hematological evaluation and the oncologist suspected of HCL. The patient was referred for an incisional biopsy in the femur region under local anesthesia, in an outpatient setting, for diagnostic confirmation. An incision was made on the right side and a tissue sample was sent for histopathological study. Histological examination of the biopsied specimens revealed bone tissue fragments that, in general, were well differentiated and without significant histological alterations, including in the cortical and medullary layers. In a few areas, however, histiocyte cell deposits with eosinophilic or foaming cytoplasm were observed. Very rarely, such cells were binucleated and exhibited folds in the nuclear membrane. There were some multinucleated giant cells, with frequent lymphoplasmocytes intermingled with groups of eosinophilic polymorphs (Figure 2). Tissue sections exhibited positive immunostaining results for S100, CD68, CD1A, CD20, CD3, but were negative for myeloperoxidase proteins.
A diagnosis of LCH was confirmed. After the histopathological examination, pharmacotherapy was initiated for the treatment and control of the disease. Sodium pamidronate (90 mg; Pamidronate, Cristalia, Brazil) was administered parenterally every 30 days, under an oncologist's supervision. After starting the medication, the patient was referred to an orthopedician who primarily suggested a bone surgery for prosthesis placement to stabilize the clinical picture; however, the patient did not consent to this surgery. Six years after the initial symptom onset, while still under the aforementioned chemotherapy, the patient presented with intense headache. She underwent an imaging examination of the skull and a scalp biopsy was performed; histological examinations of the biopsied specimens revealed sections of ulcerated skin, which showed neoplasia and infiltration of Langerhans Cells in the dermis. The cells had abundant eosinophilic cytoplasm and vesicular nucleus, were rhiniform, with a small nucleolus and characteristic longitudinal nuclear grooves. Langerhans Cells and epidermotropism were observed by the histiocyte groups.
Eosinophilic abscesses accompanied by lymphocytes were present. As decided by the oncologist, radiotherapy was started, with a cycle of three sessions. However, because of persistent pain, pulse therapy with corticoids was indicated to alleviate the painful symptomatology. From 2014 to 2016, the patient underwent a rigorous treatment protocol with 4 mg zoledronic acid (Zometa, Novartis, Brazil) administered parenterally. The disease activity was controlled, and this was verified by CBC and CRP on a monthly basis. In 2017, she presented with pain in the lower abdominal region and underwent another imaging scan, which revealed the presence of a soft tissue lesion. On this occasion, a hysteroscopy was requested, which revealed fragments of endometrial mucosa with fusocellular stroma, with areas of stromal collapse, interstitial hemorrhage, and fibrin microthrombi in the stromal vessels.
The endometrial glands showed no evidence of mitosis, vacuoles, or secretion. Edema and intense inflammatory infiltration of monomorphonuclear cells was noted in the lamina propria, with the formation of reactive lymphoid follicles and areas of accumulation of epithelial histiocytes, in addition to numerous plasmocytes and some eosinophils.The patient underwent a surgery for removal of the cervical wall lesions. Since 2017 to date, the clinical symptoms of LCH have been effectively controlled with monthly administration of parenteral zoledronic acid (4 mg; Zometa, Novartis, Brazil). Currently, the patient is on follow-up with an oncologist. During all the years of treatment for the clinical condition, the patient reported no dental follow-up, but only routine consultations for low-complexity procedures. Any lesions on oral mucosa were documented.
Discussion
LCH is defined as a clonal disorder of monomorphonucleated cells that can affect bone, skin, and mucosa. Because of the broad spectrum of clinical manifestations and the extreme diversity of disease, LCH remains a diagnostic dilema [5]. The presente case report describes a LCH with multiple lesions. The patient presented with painful symptoms at age of 20-years. Although a predilection for gnathic bones was reported in 20% of the cases, in this patient, the syndrome primarily involved the skull, femur and iliac bones as well as skin and uterus. All these locations are reported to be commonly affected in the vast majority of cases described in the literature [2,11].
To date, the exact underlying pathogenesis of this condition in one or more bones of the human body remains unknown. It is hypothesized that the immune system can modulate the location of the lesion sites [12]. Recently, De Menthon, et al. documented the existence of a mutation in the BRAVFV600E gene, which is responsible for cellular activation and transduction, through RAS, MAF and MERK pathways that have been related to LCH and cancer occurrence [13-15].
LaLitha, et al. [9] reported that LCH with solitary lesion can be considered as a single or multiple bone lesion that does not present with visceral involvement [10]. In the present case report, the patient presented not only lytic bone lesions but also skin and uterus lesions. Then, even without clinical evidence of exophthalmia and diabetes insipidus, this case report was classified as HCL with multiple lesions. Histopathological examinations confirmed diagnosis of LCH with multiple lesions. As a matter of fact, morphological identification of LCH cells and positive immunochemical staining with CD1a and S100 are necessary for a diagnosis, but the immunostainig with CD207 (Langerin) is essential for a definitive one. However, in this case, the basic pannel of antibodies and the clinical and radiographic profile of the patient were enough to recognize LCH [7].
By transmission electron microscopy, it is often possible to visualize the cytoplasmic granules-also called Birbeck granules-in the histiocytes of patients. However, for this patient, it was not possible to submit the excised tissue for this ultrastructural analysis, although it is known that the verification of the granules confirms the histopathological diagnosis established by conventional light microscopy. An immunohistochemical study of the tissue for S100 and CD1A proteins, which present with predominantly membrane immunostaining, has been described in the literature as being decisive for diagnostic confirmation [1,7,13].There are some oral alterations whose occurrence may be related to this condition. Deep periodontal pockets, alveolar bone destruction, mobility, and tooth loss are the most common and expected manifestations in patients with this disease [6,11]. These findings may occur in the form of single or multiple lesions. The cortical bone may be affected, causing mucosal ulcerations, gum inflammation, recession, and necrosis [7]. However, in the present case, no noteworthy oral changes were reported by the patient. A possible hypothesis is that the systemic changes impacted the patient's quality of life, and that the symptoms were debilitating enough for the patient to neglect her oral health. The role of dentists is extremely important in the follow-up of patients with LCH because it is possible to detect early clinical signs that indicate the presence of the disease, such as spontaneous gum bleeding, which can mimic gingivitis caused by other etiologies [7]. Additionally, in cases like the one reported, it is necessary to suggest complementary imaging tests for the maxilla and mandible such as digital radiographs.
In view of the clinical, radiological, and histopathological findings described, the patient was referred for bisphosphonate therapy. The literature states that the molecular activity of this drug in its mechanism of action during treatment is still not well-defined, although it is known to act specifically on bone metabolism, inhibiting osteoclastic resorption [7]. It is also suggested that the administration of corticosteroids, such as prednisolone, in combination with antineoplastics, such as vinblastine, may be effective, as long as the extent of the lesion is evaluated [15]. In the present clinical case, the patient was intravenously administered monthly doses of bisphosphonates, which resulted in adequate control of bone activity.
Final Considerations
This report describes a clinical case LCH with multiple lesions. Although different anatomical sites were affected in this patient, no oral lesion was found. Furthermore, although the patient presented a good clinical course after using osteolytic inhibitor drugs prescribed by the oncologist, it is suggested that the inclusion of the dentist in the multiprofessional health team should be encouraged for the early diagnosis of clinical and imaging signs of this disease in the oral cavity.
References
- Bacarin JV, Velho PHA, Slovinski AP, et al. (2014) Histiocitose de celulas de langherhans histiocitose de celulas de langherhans. relato de caso langerhans cell histiocytosis case report. Rev Med: 67-70.
- Neville BW, Allen CM, Damm DD (2009) Disturbios hematologicos. In: Neville BW, Allen CM, Damm DD, Bouquot JE, Patologia: Oral & Maxilofacial. Rio de Janeiro, Elsevier, 592-594.
- Abla O, Rollins B, Ladisch S (2019) Langerhans cell histiocytosis: Progress and controversies. Br J Haematol 187: 559-562.
- (2020) WHO classification of tumours editorial board. WHO classification of tumours of soft tissue and bone. (5th edn), IARC Press, Lyon, France.
- Howarth D (1999) Langerhans cell histiocytosis diagnosis, Natural History, Management, and outcome. Cancer 85: 2278-2290.
- Cisternino A, Asa'd F, Fusco N, et al. (2015) Role of multidisciplinary approach in a case of langerhans cell histiocytosis with initial periodontal manifestations. Int J Clin Exp Pathol 8: 13539-13545.
- Silva RN, Fernandes DT, Fonseca FP, et al. (2018) Oral manifestations of langerhans cell histiocytosis: A case series. Spec Care Dentist 38: 426-433.
- Phulware RH, Guleria P, Iyer VK, et al. (2019) Cytological diagnosis of langerhans cell histiocytosis: A series of 47 cases. Cytopatholgy 30: 413-418.
- LaLitha Ch, ManjuLa M, SriKant K, et al. (2015) Hand schuller christian disease: A rare case report with oral manifestation. J Clin Diagn Res 9: 28-30.
- Lichtenstein L (1694) Histiocytosis X eosinophilic granuloma of bone, letterer-siwe disease, and schiiller-christian disease further observations of pathological and clinical importance. J Bone Joint Surg 46: 76-90.
- Dantas MVM, Souza PBRN, Gabrielli MAC (2019) Diagnosis and management of eosinophilic granuloma of the jaw: A case report. Rev Gauch Odontol 67: 1-6.
- Rapp GE, Motta ACF (2000) Doença periodontal associada à histiocitose de celulas de langerhans. Relato de um caso clinico. Braz Dent J 11: 59-66.
- Alpizar JLQ, Monge RB (2012) Histiocitosis de celulas de langerhansenhueso parietal. Med leg Costa Rica 29: 97-101.
- De Menthon M, Meignin V, Mahr A, et al. (2017) Histiocitose decelulas de langerhans adultas. La Presse Medicale 46: 55-69.
- Allen CE, Merad M, McClain KL (2018) Langerhans-cell histiocytosis. N Engl J Med 379: 856-868.
Corresponding Author
Alena Ribeiro Alves Peixoto Medrado, Department of Oral Pathology, Bahiana School of Medicine and Public Health, Salvador, Bahia, Brazil
Copyright
© 2020 De Oliveira GC, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.