Loss of Displaced Starburst Amacrine Cells in a Rat Glaucoma Model
Abstract
Acetylcholine (ACh) release by starburst amacrine cells (SACs) has been implicated in the production of retinal waves during early retinal development. Inhibitory gamma-aminobutyric acid (GABA) release by SACs is known to be involved in directional selectivity in the mature retina. However, the role of SAC release of ACh in the adult developed mammalian retina is not entirely understood. Some evidence suggests a neuroprotective effect on retinal ganglion cells (RGCs) acting through alpha7 nicotinic acetylcholine receptors (nAChRs) present in RGCs. If ACh released by SACs is neuroprotective to RGCs, it would follow that this cholinergic transmission might be compromised in glaucoma conditions, at a time point prior to RGC loss. In this study, hypertonic saline injections into the episcleral veins surrounding the eyes of adult Long Evans rats were used to induce glaucoma-like conditions. The effect of this rat glaucoma model on starburst amacrine cell populations was examined at various time points post-insult. Additionally, ACh content within the retina and the expression of nAChRs within the retina were analyzed. It was found that starburst amacrine cell numbers, ACh content, and alpha7 nAChR expression all began to decrease one week after the procedure to induce glaucoma-like conditions, preceding the significant loss of RGCs that typically occurs one month after the same procedure. In pharmacological studies, DMP-543 and Donepezil were used to enhance the effect of ACh at the amacrine cell-RGC synapse. Each of these agents increased RGC survival in the induced glaucoma model. The results obtained from this study support the hypothesis that SACs mediate cholinergic neuroprotection for RGCs. This indicates a potentially novel role of ACh released from SACs in the mature mammalian retina.
Keywords
Glaucoma, Starburst amacrine cell, Rat, Acetylcholine, Retinal ganglion cell, Neuroprotection
Abbreviations
AC(s): Amacrine Cells; ACH: Acetylcholine; AF488: Alexa Fluor 488; AF594: Alexa Fluor 594; ChAT: Choline Acetyltransferase; CNS: Central Nervous System; CO2: Carbon Dioxide; DMSO: Dimethyl Sulfoxide; DS: Directionally Selective; ELISA: Enzyme-Linked Immunosorbent Assay; FBS: Fetal Bovine Serum; GABA: Gamma-Aminobutyric Acid; GCL: Ganglion Cell Layer; HPLC: High Performance Liquid Chromatography; IACUC: Institutional Animal Care and Use Committee; INL: Inner Nuclear Layer; IPL: Inner Plexiform Layer; IOP: Intraocular Pressure; KAX: Ketamine Acepromazine Xylazine cocktail; LC/MS/MS: Liquid Chromatography/Mass Spectrometry/Mass Spectrometry; mAChR(s): Muscarinic Acetylcholine Receptor(s); mRNA: Messenger RNA; nAChR(s): Nicotinic Acetylcholine Receptor(s); NaCl: Sodium Chloride; NO: Nitric Oxide; NRF2: Nuclear Factor Erythroid-2 Related Factor; NTG: Normal Tension Glaucoma; ONH: Optic Nerve Head; PBS: Phosphate Buffered Saline; PFA: Paraformaldehyde; RGC(s): Retinal Ganglion Cell(s); ROS: Reactive Oxygen Species; SAC(s): Starburst Amacrine Cell(s); SPE: Solid Phase Extraction; TNF: Tumor Necrosis Factor
Introduction
Glaucoma is a group of degenerative retinal diseases characterized by progressive loss of retinal ganglion cells (RGCs) and their axons that make up the optic nerve leading to irreversible loss of vision. As a leading cause of irreversible blindness in the world, it is estimated that there will be 79.6 million patients with glaucoma by 2020 [1]. Currently, the cause of glaucoma is not completely understood and no cure has been identified. The primary risk factor of glaucoma is an increase of intraocular pressure (IOP) and all current treatments are designed to lower IOP [2-4]. However, these treatments alone are often unable to stop the progression of vision loss associated with this disease. Even after successful IOP reduction, RGCs can continue to die [5-7]. Successful future glaucoma treatments will likely involve a combination of IOP reduction and neuroprotective treatments.
Neuroprotection has been defined as any intervention that prevents optic nerve damage or RGC death [8]. However, the development of neuroprotective treatment options provides a challenge because of the wide range of suspected pathological processes involved in the loss of RGCs associated with glaucoma. The neurotransmitter, acetylcholine (ACh), has been linked to neuroprotection against excitotoxic cell death and neurodegenerative diseases of the central nervous system (CNS) [9-15]. Results from our lab have demonstrated that acetylcholine provides neuroprotection against glutamate-induced excitotoxicity in isolated pig and rat RGCs in vitro [16-18]. Additional studies using an in vivo rat model of glaucoma have shown that intravitreal injections or eye drop application of the α7 nAChR selective agonist, PNU-282987, triggers neuroprotection against the loss of RGCs normally associated with glaucoma through activation of α7 nAChRs [19,20].
A specific population of cells in the retina, the starburst amacrine cells (SACs), are known to release ACh in the mammalian retina [21]. Amacrine cells are interneurons of the retina that make synapses with ganglion cells as well as bipolar cells in the inner plexiform layer (IPL). There are an estimated 40 types of amacrine cells in the vertebrate retina based on morphology, synapses and chemical messengers they contain [22]. Different types are classified by the width of their receptive fields, their stratification patterns, and by the type of neurotransmitter they release. Of these many types of amacrine cells, SACs are the only type known to release acetylcholine. There are two known populations of SACs. ACh-a-type starburst amacrine cell bodies are located in the INL and their dendrites stratify to stratum 2 of the IPL. The focus of this study was on displaced amacrine cells (ACh-b type), whose cell bodies lie in the GCL and whose dendrites stratify in strata 4 of sublamina b in the IPL. Displaced SACs synapse directly onto RGCs [22-25].
During early retinal development, SACs release ACh, which is necessary for the production of retinal waves [26,27]. In addition to ACh, SACs also release inhibitory gamma-aminobutyric acid (GABA) in the vertebrate retina onto nAChRs of directionally selective (DS) RGCs [26-32]. However, the role of ACh released from SACs in the developed, mature retina is unclear [27,33].
If ACh is neuroprotective to RGCs in vitro [16,18] and SACs are known to release ACh onto this receptor in RGCs of the mammalian retina, do SACs provide endogenous neuroprotection to RGCs in the adult vertebrate retina through the release of ACh? What happens to this transmission of ACh from displaced SACs under glaucoma conditions when RGCs die? Multiple studies investigating starburst amacrine cell survival using various glaucoma models have reported inconclusive and opposing results [34-36].
This study analyzes changes that occur in the cholinergic synapse between SACs and RGCs in glaucomatous conditions induced by hypertonic saline injections to the episcleral vein. Using this well-developed hypertonic glaucoma model in adult rats [19,20,37] our lab has measured significant RGC loss at one month after surgery that is correlated with an increase of intraocular pressure [19]. However, the effects of this glaucoma model on displaced SAC populations have not been analyzed. In this study, it is proposed that the transmission of ACh from SACs onto RGCs provides endogenous neuroprotection to RGCs in the mammalian retina and that this transmission is compromised in glaucoma-like conditions. Therefore, it is predicted that cholinergic SACs are lost in glaucoma-like conditions induced by hypertonic saline injections to the episcleral vein. This study will also demonstrate that the loss of displaced SACs correlates with a loss of ACh and loss of RGCs at a later time point. Lastly, the application of agents to enhance the effect of ACh at the amacrine cell-RGC synapse will be shown to increase RGC numbers after the induction of glaucoma. These studies are the first to suggest a novel neuroprotective role of the cholinergic SACs in the mature mammalian retina and provide an exciting new frontier in the fields of glaucoma and neuroprotection.
Materials and Methods
Animals
All experiments were performed on male or female Long Evans rats 3-6 months of age. The rats were bred and raised in the animal facility of Western Michigan University. They were kept at 20-25 degrees Celsius under a 12 hour light/dark cycle with free access to food and water. The experimental procedures were in accordance with the IACUC protocol at Western Michigan University.
Hypertonic saline injection to induce glaucoma-like conditions
Hypertonic saline injections into the episcleral veins of rat eyes, have been shown to cause a significant loss of RGCs and a significant increase of intraocular pressure (IOP) within one month [19,37]. The injection causes blanching of the episcleral veins and scarring of the trabecular meshwork system to reduce aqueous outflow [37]. To induce glaucoma-like conditions using this procedure, adult Long Evans rats were anesthetized with an intraperitoneal injection of KAX cocktail at 0.1 ml/100 gm. KAX is made up of 5 ml of ketamine (100 mg/ml), 2.5 ml xylazine (20 mg/ml), 1 ml acepromazine (10 mg/ml), and 3 ml sterile water. When loss of withdrawal reflex after toe pinch was confirmed, the whiskers were trimmed and a topical anesthetic of proparacaine hydrochloride was applied to the eye to prevent reflex activity. A hemostat was then used to clamp the tissue surrounding the eye to bulge the experimental eye. In each animal, only one eye was injected with hypertonic saline to induce glaucoma. The other eye was left untreated and acted as an internal control. The episcleral vein of each experimental eye was then injected with 50 μl of 2M NaCl using a beveled glass microinjection needle [19]. Noticeable blanching of the episcleral vein was used to confirm successful injection [19]. In some control experiments, the hemostat was used to bulge the eye in untreated control eyes as well as in eyes that underwent the procedure to induce glaucoma-like conditions. The clamping of the hemostat had no aberrant stretching effect on the optic nerve that affected experimental outcomes. After the procedure, topical antibiotic was applied to experimental eyes and animals were monitored until full consciousness was restored. The animals were then returned to the animal facility until sacrificed at designated time points for immunohistochemical analysis.
Retina removal and preparation
At various time points after the procedure to induce glaucoma-like conditions, animals were sacrificed by CO2 asphyxiation. Rat eyes were then enucleated and the retinas were removed. Whole retinas were laid flat by cutting four small incisions in the retina to form four quadrants radiating from the optic nerve head (ONH). Retinas were pinned flat with the RGC layer facing up with cactus needles in sylgard coated petri dishes [19]. Retinas were then fixed overnight with 4% paraformaldehyde at room temperature.
Immunohistochemistry
RGCs were labeled with antibodies against the cell specific marker that labels Thy 1.1 glycoprotein in the plasma membrane [38]. SACs were identified with an antibody against the enzyme choline acetyltransferase (ChAT) responsible for synthesizing ACh. SACs are the only cells in the vertebrate retina capable of synthesizing ACh. Therefore, this antibody is commonly used to specifically identify populations of SACs. In some experiments, apoptotic cells were labeled with an antibody against cleaved caspase-3. Briefly, fixed and flat-mounted retinas were permeabilized and blocked with 1% Triton-X 100 with 1% FBS in PBS for an hour and 45 minutes at room temperature. Next, fixed retinas were submerged in 0.1% Triton X-100 in PBS for 5 minutes. Retinas were then rinsed twice in PBS for 5 minutes. Finally, each retina was incubated in mouse primary antibody anti rat-Thy 1.1 (1:300, 0.5 mg/mL, BD Pharmingen 554892), sheep anti rat-ChAT (1:5000, 0.5 mg/mL, ABCAM Ab18736), or rabbit anti-rat cleaved caspase-3 (Asp175) (1:300, 10 mM, Cell Signaling Technology #9661) overnight at room temperature. The following day, after rinsing, retinas were incubated in secondary antibody with either goat anti-mouse Alexa Fluor 594 (1:300, 2 mg/mL, Life Technologies, A11005) and donkey anti-sheep Alexa Fluor 488 (1:300, 2 mg/mL, Life Technologies, A11015) or donkey anti-rabbit Alexa Fluor 594 (1:300, 2 mg/Ml, Life Technologies, A21207) overnight at room temperature. The following day, retinas were liberally washed with PBS and mounted on microscope slides with 50% glycerol/50% PBS for viewing.
Confocal microscopy and data analysis
All stained retinas were visualized with a Nikon C2+ scanning laser confocal microscope. Using the z-stack acquisition function of the microscope, a minimum of four high resolution z-stacks were obtained, 4 mm from the ONH, from each of the 4 quadrants of each retina [19,20]. Image J software was then used to visualize each z-stack and allow the counter to scroll beneath axon bundles found in the nerve cell layer. A fixed 200 × 200 μm2 grid was applied to all images and the Thy 1.1-positive RGCs within the grid for each retinal quadrant were counted blindly. In the same manner, ChAT-positive SACs were counted. The counts from the 4 quadrants were averaged for each retina. The average number of Thy 1.1-positive RGCs and ChAT-positive SACs in a retina represented an N of 1.
IOP measurements
IOP measurements were obtained daily from awake rats before and after hypertonic injections into the episcleral veins using a rebound tonometer (Mentor 0 & 0, Inc) [19,20,39]. Briefly, animals were removed from their home cages and held gently. A handheld rebound tonometer was then applied to experimental eyes to obtain IOP measurements before and after hypertonic injections were performed. On each day, 3 IOP measurements were obtained from each experimental animal and averaged. This was performed for between 8 and 20 animals depending on designated end time point specified by experimental protocols.
LC/MS/MS analysis
At the designated time points after glaucoma-inducing surgery, animals were sacrificed by CO2asphyxiation. Retinas were removed, rinsed in PBS, weighed, and immediately packaged in dry ice for delivery to the Michigan Innovation Center of Kalamazoo, MI for LC/MS/MS analysis and quantification of ACh. LC/MS/MS was performed on a Waters Quattro Micro triple quadrupole mass spectrometer using positive ion electrospray ionization. A Waters CapLC capillary HPLC was configured for on-line SPE. Each sample was done in triplicate. ACh concentration data was obtained for each sample from the Michigan Innovation Center of Kalamazoo after producing a standard curve. The average for each experimental condition was calculated and compared to controls to determine if ACh levels decrease after inducing glaucoma and to determine if this loss correlates to the loss of ChAT-positive cells.
ELISA assay
Following glaucoma inducing surgery, animals were sacrificed at designated time points. All retinas were removed and extensively homogenized for approximately 3 minutes using a plastic pestle and homogenization tube (Kimble-Chase #749521-1590) in 500 μL PBS, according to kit instructions. Then, retinas were centrifuged at 5000 rpm for 15 minutes. The supernatant was collected and serial dilutions were made. Quantification of α7 nAChRs in each serial dilution was performed using the rat neuronal acetylcholine receptor subunit alpha-7 ELISA kit from MyBioSource (Cat: MBS9340394) according to kit instructions using a generated standard curve. Measurements of absorbance at 450 nm were obtained with a BioTek Epoch microplate reader. The average absorbance was calculated for each sample condition and the absorbance of the blank was subtracted to give the adjusted value. The adjusted absorbance was then used to calculate the average optical density in terms of concentration in ng/gm of retinal tissue.
Agonist administration
For three days prior to glaucoma induction, rats received once daily eye drops of either DMP-543 (TOCRIS Bioscience, Cat No. 2330) or Donepezil (Sigma-Aldrich, D6821) at various concentrations in the right eye only. The left eye in each animal acted as an untreated internal control. The following day, the right eye of each animal was induced with glaucoma as outlined previously. Each animal then received specified eye drops once daily for two weeks after glaucoma induction. All animals were sacrificed at four weeks after the procedure to induce glaucoma. Retinas were removed, fixed in 4% paraformaldehyde overnight at room temperature, flat-mounted, and stained with an antibody against Thy 1.1 as described above. Stained retinas were then analyzed for RGC quantification. Quantification took place in the same manner previously described for quantification of ChAT-positive SACs, except that Thy 1.1-positive RGCs were counted.
mRNA sequencing
Animals were injected with hypertonic saline to induce glaucoma-like conditions as outlined above and sacrificed 3 days later by CO2 asphyxiation. Upon immediate retinal removal, an RNEasy Plus Mini Kit (Qiagen, Cat No. 74134) was used to extract total RNA according to kit instructions. Total RNA yield was verified using a Thermo Scientific nano drop 2000 spectrophotometer. Samples were then sent on dry ice to GENEWIZ, Inc. for differential gene expression analysis using Illumina HiSeq technology as well as bioinformatics analysis. Gene hit counts and expression values were calculated, transformed, and normalized. The fold change was calculated between different samples and pair-wise comparisons were generated. Pathway enrichment analysis was then performed on selected genes known to be involved in cell survival pathways and analyzed for up or down fold changes in expression.
Statistical analysis
Statistical analyses were performed using one way ANOVA in all experiments with Tukey post-hoc analysis. P ≤ 0.05 was considered statistically significant for all analyses. Cell counts from experimentally treated retinas were compared to cell counts obtained from untreated control retinas. P ≤ 0.05 represented significance from untreated controls.
Results
Time-dependent effect of glaucoma-inducing surgery on starburst amacrine cell and retinal ganglion cell survival
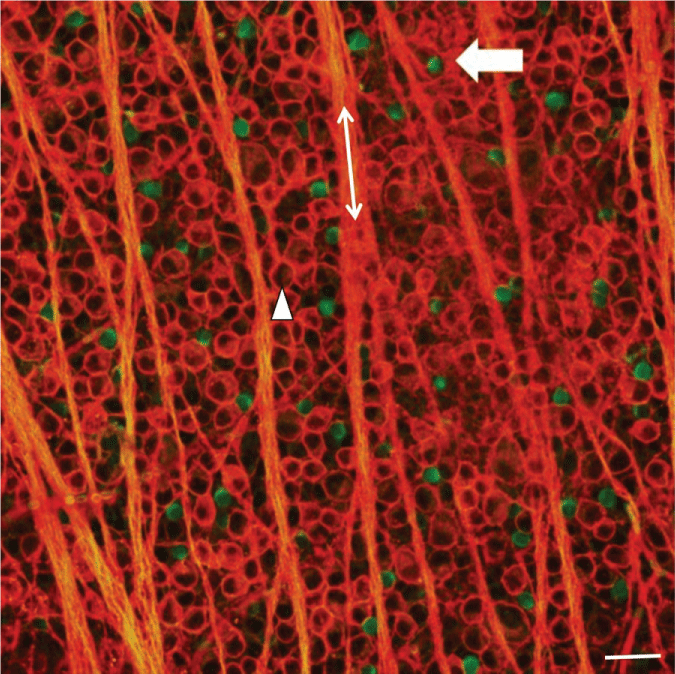
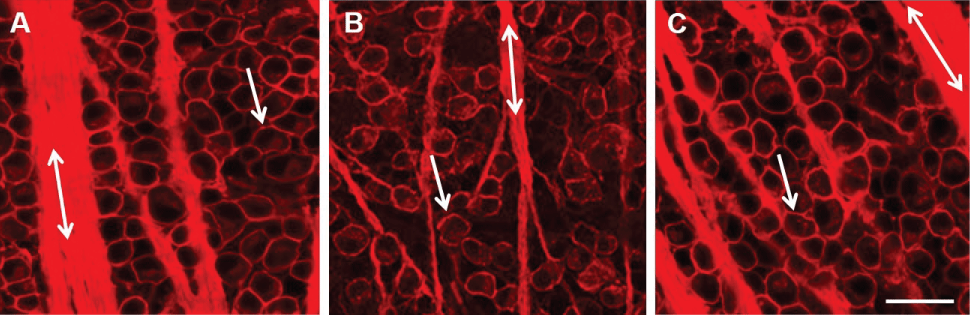
To assess the effect of induced glaucoma on SAC numbers, immunohistochemical analysis was performed on retinal flat mounts 3 days, 1 week, 2 weeks, 3 weeks, or 4 weeks after the procedure to induce glaucoma-like conditions. Retinas were double stained with antibodies against the RGC marker, Thy 1.1, and the starburst amacrine cell marker, ChAT and visualized using fluorescent secondary antibodies. Figure 1 illustrates a confocal image of a double-stained control untreated retina taken 4 mm from the optic nerve head (ONH). The arrow head points to a labeled RGC body (red), whereas the double ended arrow lies along a RGC axon fascicle (red). Displaced starburst amacrine cells are evenly distributed throughout the GCL and labeled green (arrow).
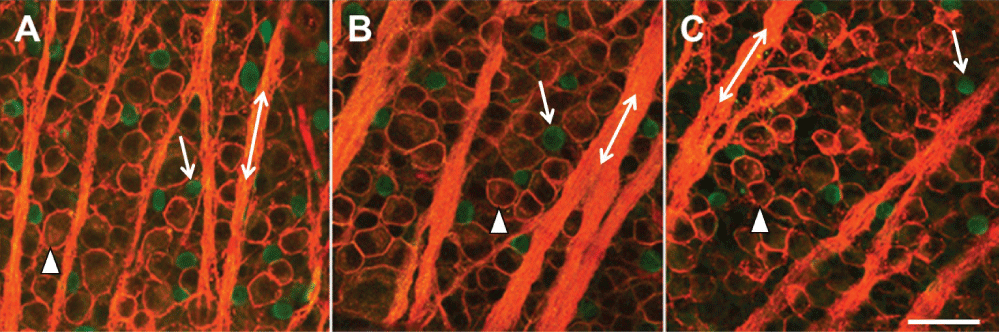
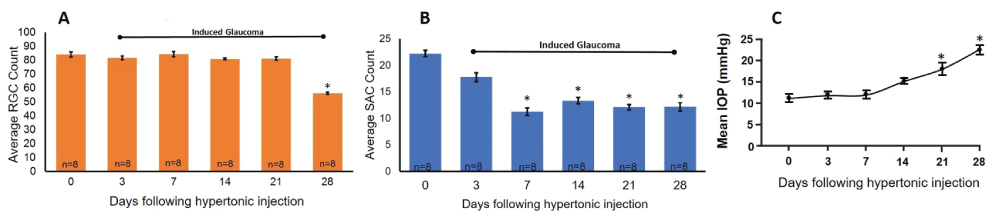
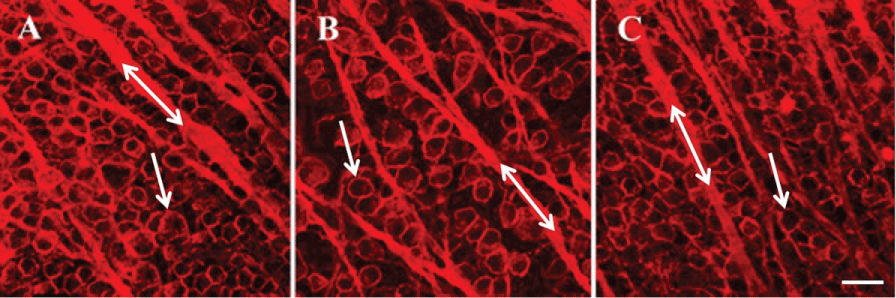
In Figure 2, images were obtained using confocal microscopy under control untreated conditions (Figure 2A), one week (Figure 2B), and four weeks (Figure 2C) after inducing glaucoma with hypertonic saline injections. Four weeks after the procedure, there was a loss of RGC bodies (arrow head) (Figure 2C and Figure 3A). Quantification of stained ChAT-positive cells from the confocal images indicated that a significant decrease in SAC number occurred as early as one-week post-insult, as compared to untreated control retinas from the same animal (Figure 2 and Figure 3). This significant loss persisted through the later time points and is summarized in Figure 3. RGC ANOVA (F(5, 90) = 67.2569, p = 6.7E-29) SAC ANOVA (F(5, 84) = 31.3123, p = 7.4E-18).
IOP measurements
IOP measurements were taken to assess the successful induction of glaucoma-like conditions in experimental eyes. The results of the obtained averaged IOP measurements are summarized in Figure 3C. Before inducing glaucoma-like conditions, the mean IOP measured in adult awake Long Evans rats equaled 11.5 mmHg (± 2.5 n = 8). A steady increase in IOP was measured across the time points measured. A significant increase from the internal control eye was measured 21 days after the induction of glaucoma-like conditions with an average IOP measurement of 17.5 mmHg (± 3.3 n = 8) and increased to an average of 22.2 mmHg (± 2.5 n = 8) 29 days after the hypertonic saline injection procedure. These results show that hypertonic saline injections used to induce glaucoma-like conditions were successful in increasing IOP in experimental eyes.
Caspase expression
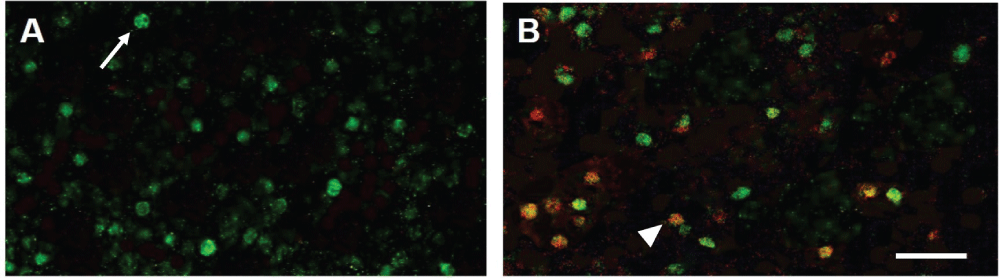
To provide evidence that the procedure to induce glaucoma in adult rats involves apoptosis of SACs, glaucomatous retinas were double-stained with antibodies against ChAT and cleaved caspase-3. Figure 4A shows a control untreated retina where ChAT-positive SACs (green) are seen (arrow). No evidence of caspase-3 (red) can be found. Figure 4B shows caspase-3 positive cells 3 days after hypertonic saline injections. Some ChAT-positive SACs are double labeled with the antibody against caspase-3 (orange; arrowhead). These results support the hypothesis that SACs begin to undergo apoptotic cell death as early as 3 days after glaucoma-inducing surgery.
Acetylcholine content
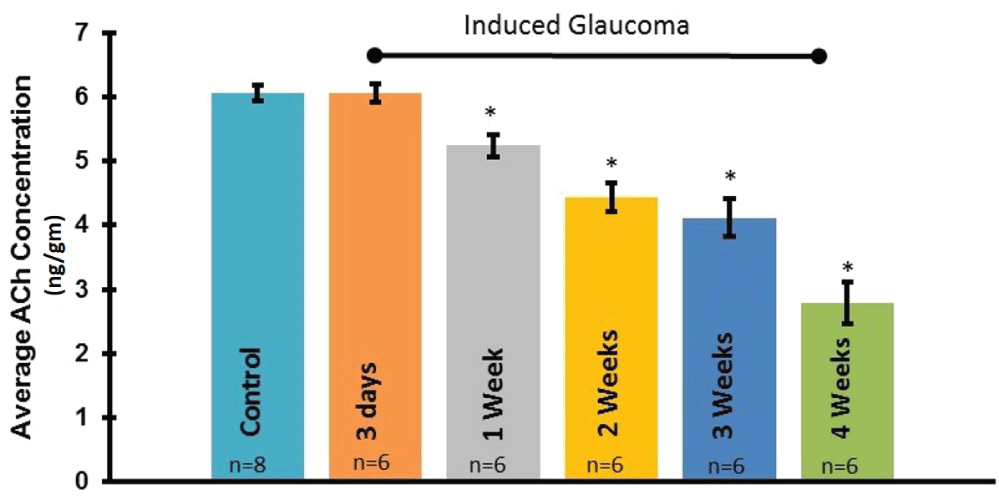
To determine if the loss of starburst amacrine cell numbers correlates with a loss of ACh content, LC/MS/MS analysis was performed on whole retinas. Figure 5 summarizes the average ACh content obtained from adult rat retinas analyzed at multiple time points before and after the procedure to induce glaucoma. The results demonstrate a significant loss of ACh in the retina as early as one week post-procedure, as compared to the untreated control condition. ANOVA (F(5, 18) = 43.2863, p = 2.1E-09). In addition, ACh content decreased in a time dependent manner between 1 and 4 weeks after the procedure to induce glaucoma-like conditions (Figure 5).
Receptor protein expression
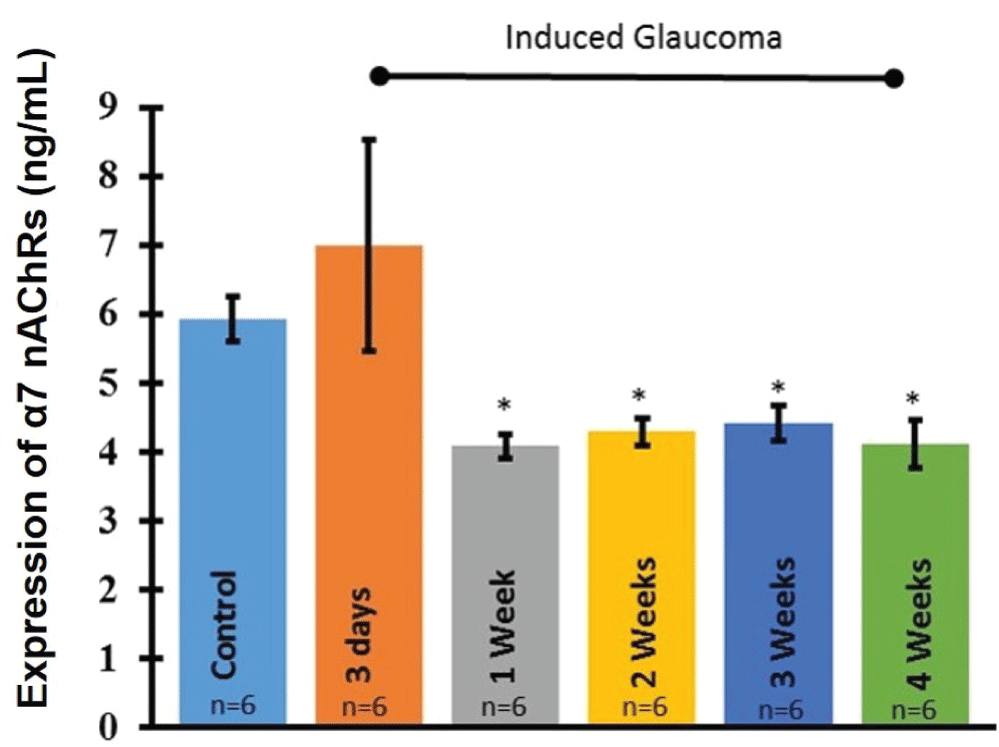
In order to examine the changes in α7 nAChR expression after glaucoma induction, ELISA analysis was performed on whole retinas under each experimental condition according to EKISA kit instructions. ELISA analysis revealed that significant loss of the α7 nAChRs occurred within one week of the procedure to induce glaucoma (Figure 6). This significant loss was maintained through all later time points. ANOVA (F(5, 6) = 5.66642, p = 0.02839).
Agonist treatment
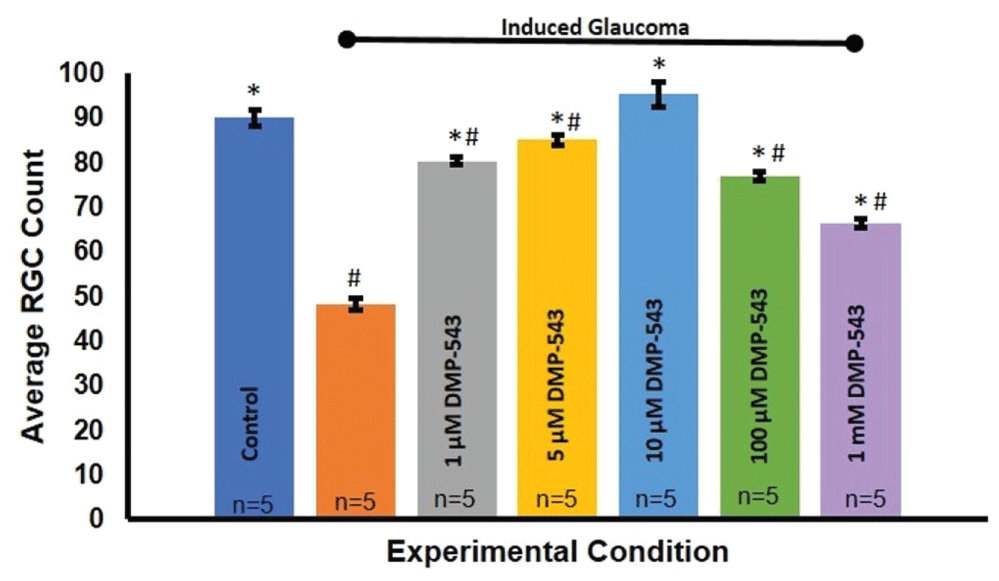
The hypothesis of this study states that ACh released from SACs within the adult mammalian retina is neuroprotective to RGCs and that this transmission is lost in glaucoma-like conditions. The results shown previously suggest that SAC transmission of ACh is indeed lost as early as one week after the induction of glaucoma. In order to determine if restoring this transmission is neuroprotective to RGCs, pharmacological agents that act on the synapse were administered as eye drop treatments. DMP-543 enhances ACh release from pre-synaptic cells [40,41]. If ACh is neuroprotective at the SAC-RGC synapse, application of DMP-543 would be expected to restore the neuroprotective effects of ACh on RGCs after inducing glaucoma. Indeed, quantification of RGC survival upon treatment with different concentrations of DMP-543 shows a dose-dependent neuroprotective effect on RGC numbers Figure 7. Figure 8 indicates that 10 μM DMP-543 achieves a maximal neuroprotective effect and prevented loss of RGCs associated with the procedure to induce glaucoma. ANOVA (F(6, 214) = 39.863, p = 2.27E-32).
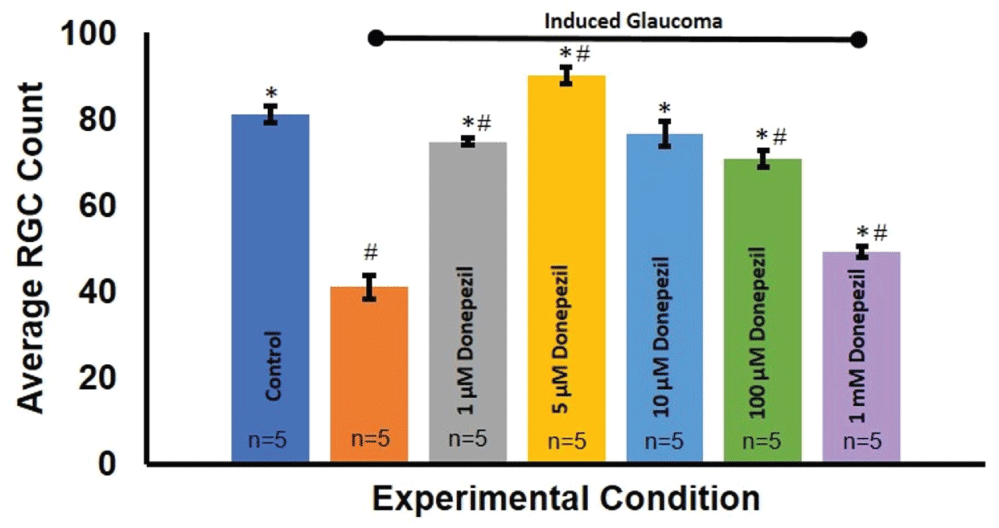
Donepezil, an acetylcholinesterase inhibitor, operates within the same synapse to prevent the degradation of ACh by the acetylcholinesterase enzyme. Similarly, with the application of Donepezil, it is expected any remaining ACh would bind to ACh receptors longer and therefore, could rescue the neuroprotective effects of endogenous ACh. Quantification studies of RGC survival before and after Donepezil treatment also revealed a dose dependent neuroprotective effect on RGC counts Figure 9. Figure 10 summarizes these effects. Specifically, 5 μm Donepezil accomplished a maximal effect on RGC survival in hypertonic injected eyes. ANOVA (F(6, 79) = 83.419, p = 4.1E-32).
α7nAChR mRNA expression
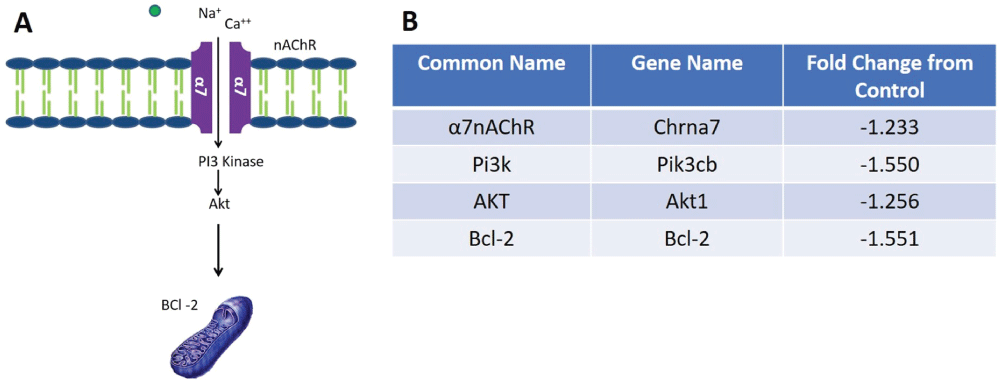
To validate the finding that α7 nAChR receptor protein expression is lost prior to the RGC death, mRNASeq analysis was conducted to observe expression of the receptor at the transcriptional level. This analysis also allowed us to investigate potential cell survival pathways that might be down regulated in the glaucoma condition compared to an untreated control condition. Fold change data revealed a 1.23 fold decrease in expression of the α7 nAChR in glaucomatous retinas compared to untreated control retinas (Figure 11) to support the hypothesis that cholinergic transmission is lost in glaucoma conditions.
Previous in vitro studies from this lab have demonstrated that activation of α7nAChRs with a selective α7nAChR agonist activates the PI3 Kinase-Akt-BCl2 cell survival pathway in isolated RGCs [42]. When pathway analysis of whole retinal mRNA transcripts was performed, a decrease in mRNA expression was found in PI3 kinase, Akt and Bcl2 when eyes were injected with hypertonic saline to induce glaucoma compared to control untreated conditions. Fold changes of mRNA expression that occurred after inducing glaucoma-like conditions are listed in Figure 11.
Discussion
The results from this study provide evidence of apoptotic loss of displaced SACs, corresponding with a reduction in ACh content and α7 nAChRs before any significant loss of RGCs occurs in an in vivo rat glaucoma model. Is the loss of SACs and reduction of ACh a factor in the eventual loss of RGCs associated with glaucoma? If so, these results provide insight into the identification of a possible new function for cholinergic amacrine cells within the adult mammalian retina. Results from this study demonstrate that cholinergic transmission is diminished prior to significant RGC loss, and that application of agents to affect ACh release and degradation at the amacrine cell-RGC synapse prevent RGC death. It is interesting to note that there was a significant loss of SACs after the procedure to induce glaucoma within 1 week but the remaining SACs remained for the duration of a month. On the other hand, ACh content significantly decreased in a time dependent manner between 1 and 4 weeks following the procedure to induce glaucoma-like conditions. This discrepancy between SAC numbers and ACh release may be due to SACs that are damaged after the procedure, which compromise the release of ACh over time. Damage to SACs is supported by caspase-3 antibody studies. Results indicate many SACs are caspase-3 positive as early as 3 days following hypertonic injections.
Further investigation into the differential expression of specific mRNA transcripts suggested down regulation of the PI3K-Akt-BCl2 cell survival pathway under glaucoma-like conditions as well a down regulation of transcripts for α7 nAChRs. This result supports previous in vitro ELISA studies performed on isolated adult mammalian RGCs that demonstrated the PI3K-Akt-BCl2 pathway was involved in neuroprotection against glutamate-induced toxicity induced by the α7 nAChR selective agonist, PNU-282987 [42]. ACh has also been implicated as a neuroprotective agent in neurodegenerative diseases. Alzheimer's disease, for instance, is a neurodegenerative disorder characterized by neuronal loss leading to brain atrophy. The activation of α7 nAChRs has been studied as a potential neuroprotective approach in Alzheimer's disease models [43] and several studies have linked neuroprotection induced by activation of α7 nAChRs and the PI3K/Akt/Bcl-2 cell survival pathway [44-46]. Activation of the α7 nAChR signaling cascade, in particular, showed significant increase in cell viability, decreased number of apoptotic cells, decreased levels of reactive oxygen species (ROS), and decreased levels of caspase-3 in rat primary cortical neurons in a neurotoxic Alzheimer's model. Blocking the receptor with the antagonist, methyllycaconitine, MLA, eliminated the neuroprotective effects [45]. It is therefore possible that thePI3K/Akt/Bcl-2 cell survival pathway identified in an in vitro RGC model of excitotoxicity is also involved in ACh-induced neuroprotection of RGCs in adult mammals in glaucoma [16,18].
Additional pathways have also been implicated in neuronal neuroprotection through the actions of the α7 nAChR. In an ischemic brain model, the activation of this receptor acted through the nuclear factor erythroid-2 related factor (Nrf2) and its target gene, heme oxygenase -1 (HO-1) to suppress cell death, ROS production, and tumor necrosis factor (TNF) release [47]. However, the model to induce glaucoma using hypertonic saline injections does not lead to ischemia and necrosis, but mimics the increase of intraocular pressure typically associated with glaucoma. As a result the procedures that induce ischemia are not likely to trigger the same pathways as the induced glaucoma model used in this study. Taken together, the results presented here provide a collection of evidence to support the hypothesis that ACh released by displaced SACs in the RGC layer of adult mammalian retina provide sretinal neuroprotection through the activation of the α7 nAChR on RGCs. When that neuroprotection is lost under glaucoma-like conditions, RGCs begin to die. These results suggest a previously unidentified role for displaced SACs and ACh in the adult mammalian retina.
Previous studies investigating starburst amacrine cell survival using various glaucoma models have reported inconclusive and even opposing results. One hereditary model of glaucoma in mice showed that amacrine cell populations were unaffected by the glaucoma condition [35]. Another study, where experimental glaucoma was induced using microbead injections demonstrated that choline acetyltransferase (ChAT)-positive and glycinergic AC subtypes were unaffected [48]. However, an ischemic/reperfusion model of glaucoma reported a substantial reduction in transcripts for the SAC-specific enzyme, choline acetyltransferase (ChAT) within 24 to 72 hours [34]. Yet another lab showed that ChAT positive amacrine cell numbers decline within five weeks of episcleral vein cauterization [36]. It is likely that conflicting results associated with these studies are due to the different methods and procedures used to induce glaucoma-like conditions. In this study, we have used hypertonic injection of 2M NaCl saline to cause vascular scar tissue and reduce outflow through the trabecular meshwork to gradually increase intraocular pressure over time. This reliable gradual increase of intraocular pressure is the primary risk factor associated with glaucoma and was preferable over models that induced ischemia [34,49,50] or that needed to inject microbeads multiple times to reduce drainage through the trabecular meshwork [48,51]. Results from this study lend support to the hypothesis that SACs are lost in glaucoma. In addition, evidence was provided that the SACs and neuroprotection provided by ACh were lost prior to RGC loss.
Studies performed with DMP-543 and Donepezil demonstrated that loss of RGCs could be prevented if the reduction of ACh associated with the procedure to induce glaucoma was supplemented with DMP-543 to enhance ACh release from remaining SACs or with Donepezil to reduce the degradation of ACh on the RGC postsynaptic membrane. These results provide evidence of the neuroprotective action of ACh. When ACh levels decrease under glaucomatous conditions, cell survival pathways are no longer activated and loss of RGC occurs. As a result, future glaucoma treatments may involve a combination of treatments that target intraocular pressure changes as well as regulation of ACh levels in the retina.
Acknowledgements
This work was supported by an NIH NEI grant (EY 022795) issued to Dr. C. Linn.
References
- Quigley HA, Broman AT (2006) The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90: 262-267.
- Chauhan BC, Pan J, Archibald ML, et al. (2002) Effect of intraocular pressure on optic disc topography, electroretinography, and axonal loss in a chronic pressure-induced rat model of optic nerve damage. Invest Ophthalmol Vis Sci 43: 2969-2976.
- Damji KF, Behki R, Wang L (2003) Canadian perspectives in glaucoma management: Setting target intraocular pressure range. Can J Ophthalmol 38: 189-197.
- Casson RJ, Chidlow G, Wood JP, et al. (2012) Definition of glaucoma: clinical and experimental concepts. Clin Exp Ophthalmol 40: 341-349.
- Cockburn DM (1983) Does reduction of intraocular pressure (IOP) prevent visual field loss in glaucoma? Am J Optom Physiol Opt 60: 705-711.
- Brubaker RF (1996) Delayed functional loss in glaucoma. LII Edward Jackson Memorial Lecture. Am J Ophthalmol 121: 473-483.
- Gogate P, Deshpande R, Chelerkar V, et al. (2011) Is glaucoma blindness a disease of deprivation and ignorance? A case-control study for late presentation of glaucoma in India. Indian J Ophthalmol 59: 29-35.
- Sena DF, Lindsley K (2013) Neuroprotection for treatment of glaucoma in adults. Cochrane Database Syst Rev CD006539.
- Marin P, Maus M, Desagher S, et al. (1994) Nicotine protects cultured striatal neurones against N-methyl-D-aspartate receptor-mediated neurotoxicity. Neuroreport 5: 1977-1980.
- Shimohama S, Akaike A, Kimura J (1996) Nicotine-induced protection against glutamate cytotoxicity. Nicotinic cholinergic receptor-mediated inhibition of nitric oxide formation. Ann N Y Acad Sci 777: 356-361.
- Kaneko S, Maeda T, Kume T, et al. (1997) Nicotine protects cultured cortical neurons against glutamate-induced cytotoxicity via alpha7-neuronal receptors and neuronal CNS receptors. Brain Res 765: 135-140.
- Dajas-Bailador FA, Lima PA, Wonnacott S (2000) The alpha7 nicotinic acetylcholine receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a calcium ion dependent mechanism. Neuropharmacology 39: 2799-2807.
- O'Neill MJ, Muaary TK, Lakies V, et al. (2002) The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurode-generation. Curr Drug Targets CNS Neurol Disord 1: 399-411.
- Gahring LC, Meyer EL, Rogers SW (2003) Nicotine-induced neuroprotection against NMDA or beta-amyloid peptide occur through independent mechanisms distinguished by pro-inflammatory cytokines. J Neurochem 87: 1125-1136.
- Nakamizo T, Kawamata J, Yamashita H, et al. (2005) Stimulation of nicotinic acetylcholine receptor protects motor neurons. Biochem Biophys Res Commun 330: 1285-1289.
- Wehrwein E, Thompson SA, Coulibaly SF, et al. (2004) Acetylcholine Protection of Adult Pig Retinal Ganglion Cells from Glutamate-Induced Excitotoxicity. Invest Ophthalmol Vis Sci 45: 1531-1543.
- Thompson SA, Smith O, Linn DM, et al. (2006) Acetylcholine neuroprotection against glutamate-induced excitotoxicity in adult pig retinal ganglion cells is partially mediated through alpha4 nAChRs. Exp Eye Res 83: 1135-1145.
- Iwamoto K, Linn DM, Mata D, et al. (2013) Neuroprotection of rat retinal ganglion cells mediated through alpha7 nicotinic acetylcholine receptors. Neuroscience 237: 184-198.
- Iwamoto K, Birkholz P, Schipper A, et al. (2014) A nicotinic acetylcholine receptor agonist prevents loss of retinal ganglion cells in a glaucoma model. Invest Ophthalmol Vis Sci 55: 1078-1087.
- Mata D, Linn DM, Linn CL (2015) Retinal ganglion cell neuroprotection induced by activation of alpha7 nicotinic acetylcholine receptors. Neuropharmacology 99: 337-346.
- Masland RH, Tauchi M (1988) The cholinergic amacrine cells. Trends Neurosci 9: 218-223.
- Kolb H, Fernandez E, Nelson R (2005) Roles of Amacrine Cells. In: The Organization of the Retina and Visual System. Webvision, Salt Lake City (UT): University of Utah Health Sciences Center.
- Famiglietti EV, Siegfried EC (1980) The amacrine cells of the rabbit retina. Invest Ophthal Vis Sci Suppl 19: 70-71.
- Famiglietti EV (1983) ON and OFF pathways through amacrine cells in mammalian retina: the synaptic connections of “starburst” amacrine cells. Vision Res 23: 1265-1279.
- Famiglietti EV (1983) Starburst' amacrine cells and cholinergic neurons: mirror-symmetric ON and OFF amacrine cells of rabbit retina. Brain Res 261: 138-144.
- Zheng JJ, Lee S, Zhou ZJ (2004) A developmental switch in the excitability and function of the starburst network in the mammalian retina. Neuron 44: 851-864.
- Masland RH (2005) The many roles of starburst amacrine cells. Trends Neurosci 28: 395-396.
- Hayden SA, Mills JW, Masland RM (1980) Acetylcholine synthesis by displaced amacrine cells. Science 210: 435-437.
- Brecha N, Johnson D, Peichl L, et al. (1988) Cholinergic amacrine cells of the rabbit retina contain glutamate decarboxylase and gamma-aminobutyrate immunoreactivity. Proceedings of the Nat Acad of Sciences of the United States of America 85: 6187-6191.
- Vaney DI, Young HM (1988) GABA-like immunoreactivity in cholinergic amacrine cells of the rabbit retina. Brain Res 438: 369-373.
- O'Malley DM, Masland RH (1989) Co-release of acetylcholine and gamma-aminobutyric acid by a retinal neuron. Proc Natl Acad Sci U S A 86: 3414-3418.
- Grzywacz NM, Amthor FR, Merwine DK (1998) Necessity of acetylcholine for retinal directionally selective responses to drifting gratings in rabbit. J Physiol 512: 575-581.
- Taylor WR, Smith RG (2012) The role of starburst amacrine cells in visual signal processing. Vis Neurosci 29: 73-81.
- Dijk F, Leeuwen SV, Kamphuis W (2004) Differential effects of ischemia/reperfusion on amacrine cell subtype-specific transcript levels in the rat retina. Brain Res 1026: 194-204.
- Jakobs TC, Libby RT, Ben Y, et al. (2005) Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol 171: 313-325.
- Hernandez M, Rodriguez FD, Sharma SC, et al. (2009) Immunohistochemical changes in rat retinas at various time periods of elevated intraocular pressure. Mol Vis 15: 2696-2709.
- Morrison JC, Moore CG, Deppmeier LM, et al. (1997) A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res 64: 85-96.
- Barnstable CJ, Drager UC (1984) Thy-1 antigen: a ganglion cell specific marker in rodent retina. Neuroscience 11: 847-855.
- Birkholz PJ, Gossman CA, Webster MK, et al. (2016) Prevention of Glaucoma-Induced Retinal Ganglion Cell Loss Using Alpha7 nAChR Agonists. J Ophthalmol and Vis Sci 1: 1003.
- Earl RA, Zaczek R, Teleha CA, et al. (1998) 2-Fluoro-4-pyridinylmethyl analogues of linopirdine as orally active acetylcholine release-enhancing agents with good efficacy and duration of action. J Med Chem 41: 4615-4622.
- Zaczek R, Chorvat RJ, Saye JA, et al. (1998) Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)- 9(10H)-anthracenone and 10, 10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J Pharmacol Exp Ther 285: 724-730.
- Asomugha C, Linn DM, Linn CL (2010) ACh receptors link two signaling pathways to neuroprotection against glutamate-induced excitotoxicity in isolated pig RGCs. J Neurochem 112: 214-226.
- Araya JA, Ramirez AE, Figueroa-Aroca D, et al. (2014) Modulation of Neuronal Nicotinic Receptor by Quinolizidine Alkaloids Causes Neuroprotection on a Cellular Alzheimer Model. J Alzheimers Dis 42: 143-155.
- Buckingham SD, Jones AK, Brown LA, et al. (2009) Nicotinic acetylcholine receptor signaling: roles in Alzheimer's disease and amyloid neuroprotection. Pharmacol Rev 61: 39-61.
- Zhang X, Wu M, Lu F, et al. (2014) Involvement of α7 nAChR signaling cascade in epigallocatechin gallate suppression of β-amyloid-induced apoptotic cortical neuronal insults. Mol Neurobiol 49: 66-77.
- Huang X, Cheng Z, Su Q, et al. (2012) Neuroprotection by nicotine against colchicine-induced apoptosis is mediated by PI3-kinase--Akt pathways. Int J Neurosci 122: 324-332.
- Parada E, Egea J, Buendia I, et al. (2013) The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal 19: 1135-1148.
- Akopian A, Kumar S, Ramakrishnan H, et al. (2016) Amacrine cells coupled to ganglion cells via gap junctions are highly vulnerable in glaucomatous mouse retinas. J Comp Neurol.
- Li JB, Lu ZG, Xu L, et al. (2014) Neuroprotective effects of bis(7)-tacrine in a rat model of pressure-induced retinal ischemia. Cell Biochem Biophys 68: 275-282.
- Schmid H, Renner M, Dick HB, et al. (2014) Loss of inner retinal neurons after retinal ischemia in rats. Invest Ophthalmol Vis Sci 55: 2777-2787.
- Weber AJ, Zelenak D (2001) Experimental glaucoma in the primate induced by latex microspheres. J Neurosci Methods 111: 39-48.
Corresponding Author
Cindy Linn, Ph.D, Professor, Department of Biological Sciences, Western Michigan University, Kalamazoo, MI 49008, USA, Tel: +269-387-5615.
Copyright
© 2017 Cooley-Themm CA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.