Papillary Glioneural Tumor Presenting with Multiple Ischemic Strokes: An Aggressive Presentation of a "Benign" Entity
Abstract
Background: Papillary glioneural tumors are infrequent neoplasms of the central nervous system, classically presenting with an indolent clinical course, rarely being related to an aggressive presentation, and not being associated with ischemic or another type of paraneoplastic phenomena. We describe the first case of this type of presentation with a literature review of the current knowledge of this entity.
Methods and results: A 16-year-old female presented with a hemorrhagic intra-axial lesion, confirmed to be a papillary glioneural tumor in the histopathological analysis, being associated with multiple ischemic cerebral and posterior fossa strokes without another discernable than the presence of a paraneoplastic syndrome, not previously reported with this type of neoplasms. A literature review is presented detailing the current knowledge about this entity and emphasizing the need for greater knowledge about its natural history, immunobiology and treatment alternatives, opening a new window for the study of this pathology and establishing the need for a strict follow-up of patients who have this kind of tumors in order to learn more about their evolution.
Conclusion: The papillary glial tumor, a rare entity having generally with a benign course, can have different presentations. Greater knowledge is needed to understand this behavior in order to optimize patient's outcomes.
Keywords
Papillary glioneural tumor, Ischemic infarct, Paraneoplastic syndrome
Introduction
Papillary glioneural tumors are infrequent neoplasms of the central nervous system. Generally, these tumors present an indolent clinical course. There are no reports in the literature that associate these lesions with ischemic phenomena that may suggest a paraneoplastic syndrome; however, we report the case of an atypical presentation in relation to multiple intracranial ischemic lesions.
Case Report
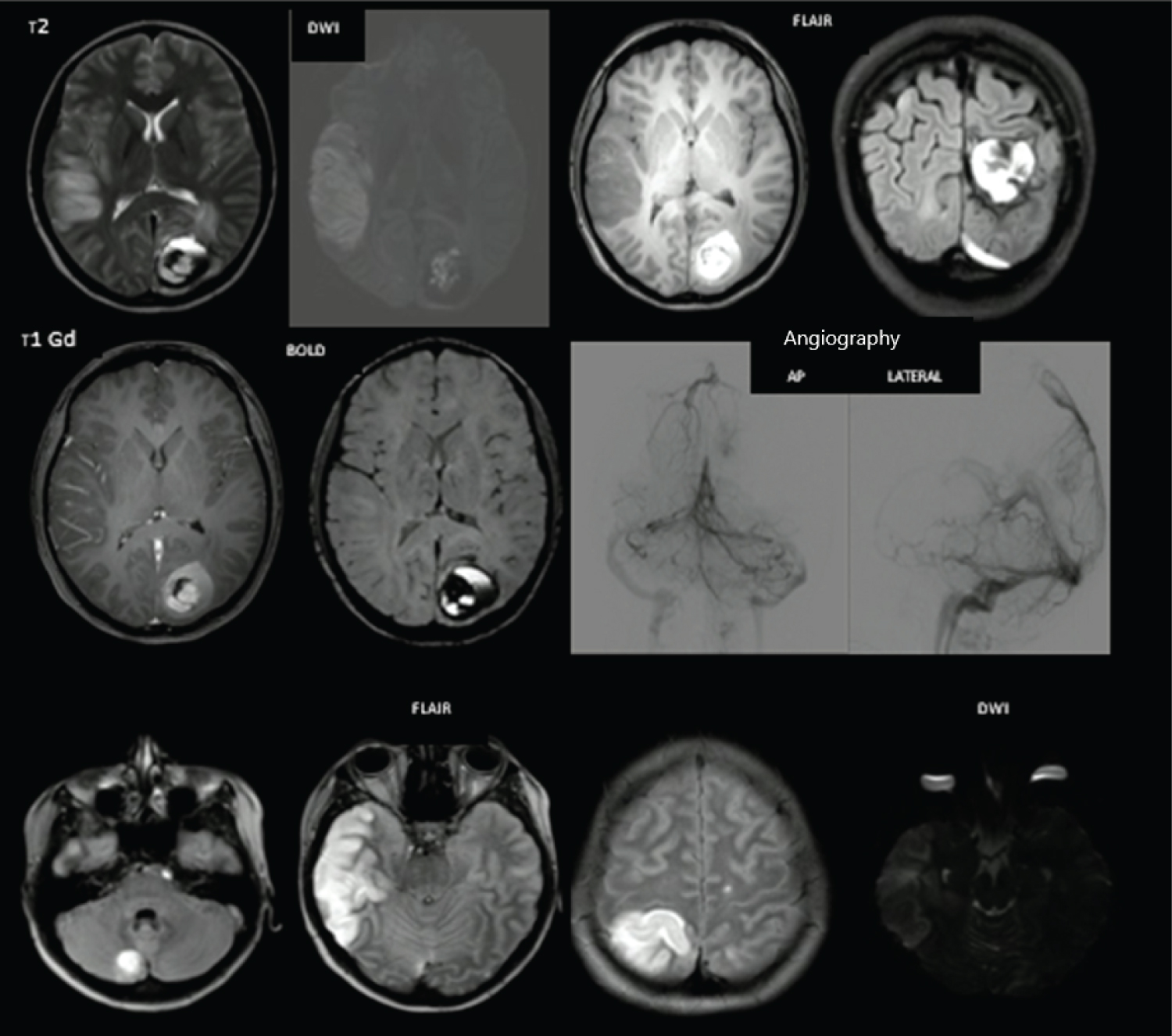
A 16-year-old female patient presented with two months of headache, asthenia, emesis and vertigo, symptoms that were markedly exacerbated on the day of the consultation. She had no relevant pathological history. The neurological examination documented limitation for abduction of the left eye, right homonymous hemianopia and left peripheral facial palsy associated with flaccid dysarthria. The patient was right-handed. Initially, a brain MRI study was requested (Figure 1) showing a predominantly subacute early hematoma in the medial and superior aspect of the left occipital lobe with a heterogeneous image inside which presented a central enhancement as well as multiple early subacute acute ischemic infarcts in both cerebral hemispheres and in the right cerebellar hemisphere. Surgical intervention was performed through a neuronavigation-guide craniotomy, performing an en bloc resection. Postoperatively, the patient was monitored in the intensive care unit without presenting an additional neurological deficit. Autoimmunity studies were carried out ruling out systemic diseases, as well as primary vasculitis of the central nervous system, with additional studies that ruled out metabolic disease, and coagulopathy workup that had negative findings. Cardiac studies had no abnormal findings, considering the ischemic phenomena in relation to the underlying tumor pathology.
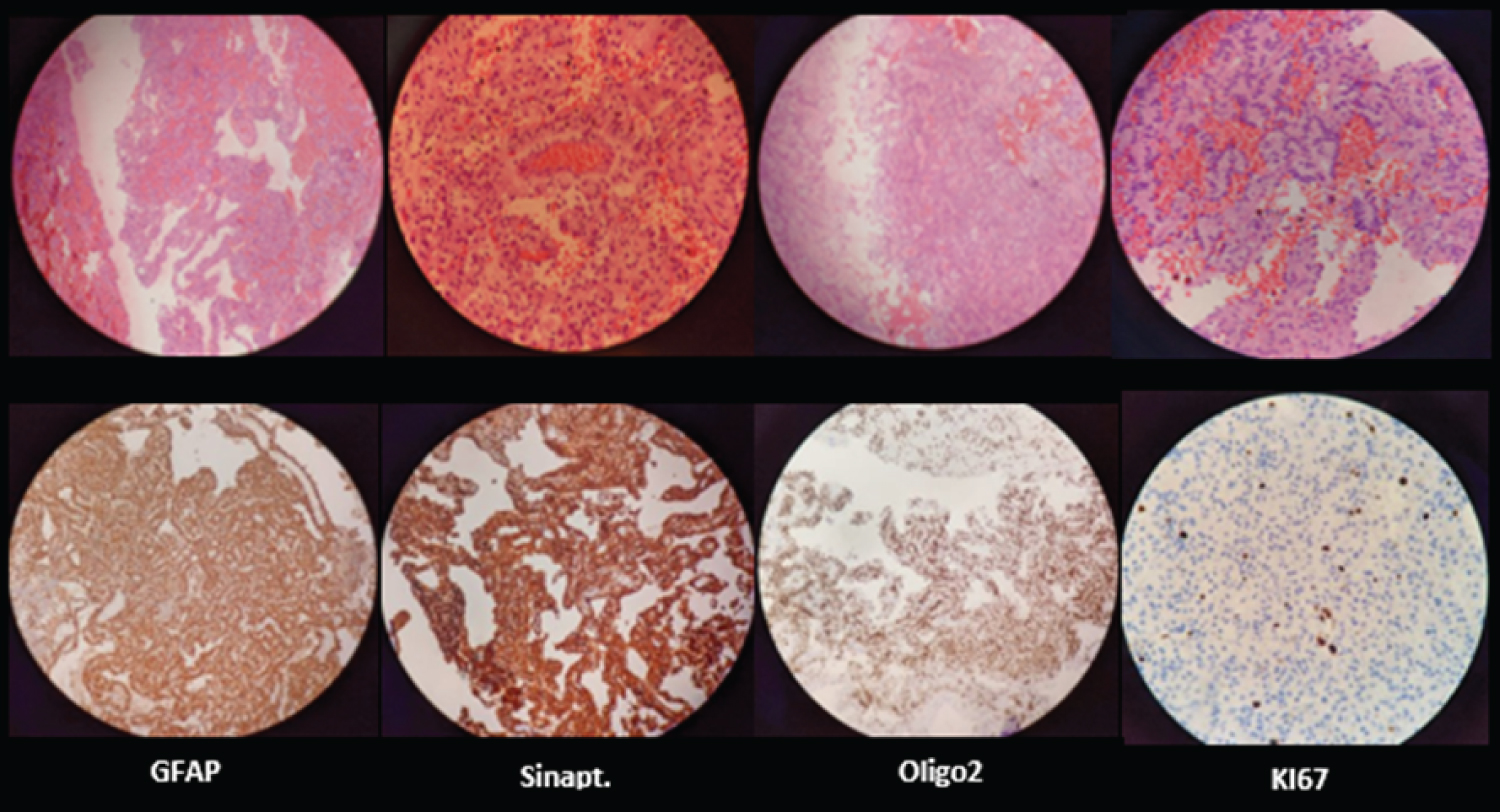
The patient was discharged home after this. The pathology study (Figure 2), in the hematoxylin-eosin sections at low magnification, showed a well-defined tumor lesion with a benign appearance, with minimal microvascular proliferation, without necrosis or cellular atypia. Glial differentiation was evidenced, as well as areas of pseudopapillary formation delineated by a single layer of cuboidal cells with scant cytoplasm and a nucleus without atypia, which surround blood vessels with hyalinized walls. Regarding cell morphology, cells with a neural appearance that formed perivascular rosettes with vascular hyalinization were evidenced. Likewise, hemosiderin deposits were identified. Immunohistochemistry showed the glial component oriented around the hyalinized blood vessels, highlighted with PAGF and OLIGO2, while the neuronal component that was distributed in the papillary area, presented neurocytic and ganglionic morphology, with marked positivity in NeuN and light with synaptophysin. The cell proliferation index (KI67) was low, being less than 2%.
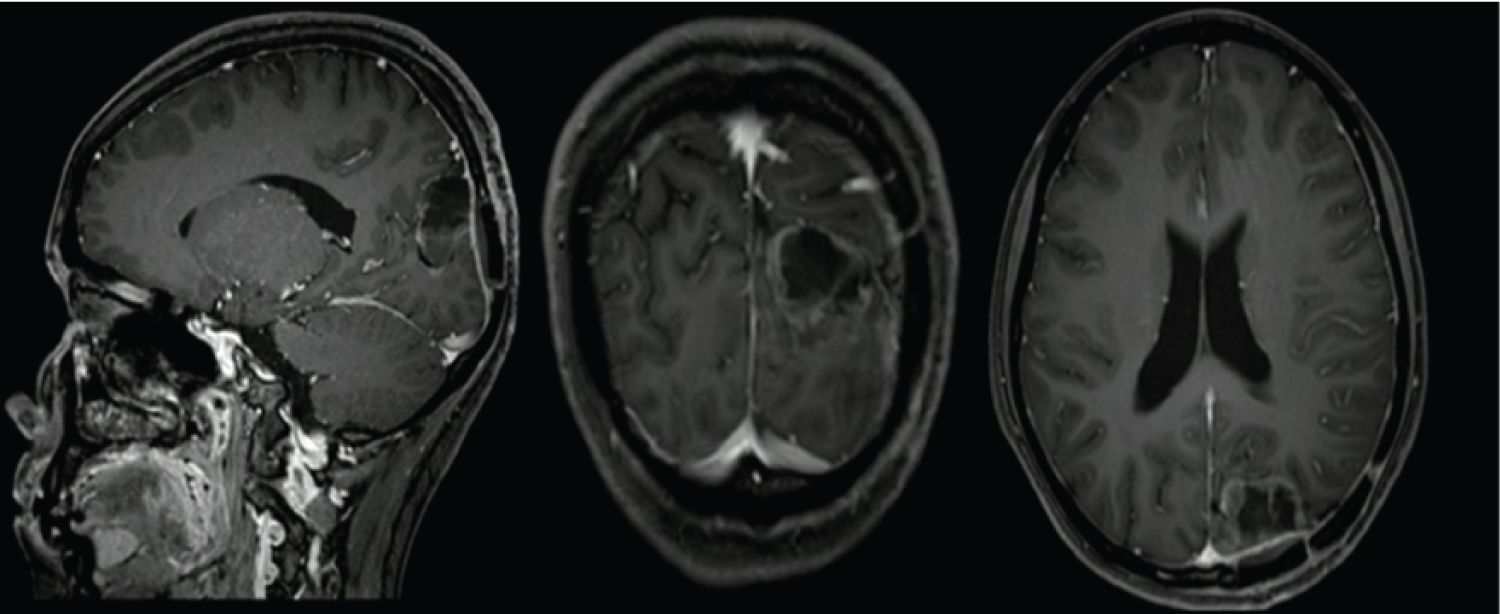
A follow-up imaging study was performed at 3 months, which ruled out residual disease (Figure 3) or recurrence, having recovery from the previously described deficit and persisting only with a known visual field compromise.
Discussion
The papillary glioneural tumor typically presents and indolent behavior and slow growth, being predominantly supratentorial, generally adjacent to the lateral ventricular system and in relation to the temporal lobe, being reported in patients from the first to the eighth decade of life (4 to 75 years) [1] and corresponding to less than 0.02% of intracranial neoplasms [2]. The first report of this pathology dates from 1998 [3], however, it was not described as a particular entity until 2007 [1].
The clinical presentation of these lesions is variable; however, the majority of patients are asymptomatic or present a scarce neurological compromise, generally having a benign clinical course [4,5]. Bleeding associated with this type of tumor is not frequent. Carangelo, et al. [1] only found two cases with hemorrhage in a series of 67 of this type of tumors, finding possibly related to the thin wall of the blood vessels in the solid region and their hyalinization in the papillary region [2].
Imaging studies characterize it as a circumscribed lesion, with a cortico-subcortical extension, or deeper in relation to the ventricular system, with cystic and solid components. Its solid components are Iso-Hypo intense in T1 and Iso-Hyper intense in T2, classically presenting a patchy or ring enhancement after gadolinium administration. They may additionally present calcifications or have a hemorrhagic component, as well as be associated with edema or mass effect if the lesions are large [6].
According to the WHO 2016 central nervous system tumor classification, this tumor is located in the section of mixed neuronal and glioneural tumors. It is characterized by a prominent pseudo papillary architecture, a single or pseudo-stratified layer of flattened or cuboidal glial cells with round nuclei and scant cytoplasm, located around hyalinized blood vessels together with intervening collections of neurocytes and medium-sized ganglion cells with accompanying neuropil [7]. In its histology, a fundus that can be fibrillary or micro cystic is described. Neurocytes frequently resemble oligodendrocytes. Towards the periphery of the tumor, scattered tumor cells intermingle with glyotic areas that may present Rosenthal fibers, eosinophilic granular bodies, hemosiderin, and micro calcifications [8,9]. It is necessary to clarify that these glial elements lack atypia and mitotic activity, likewise, microvascular proliferation and necrosis is minimal, characteristics that give this tumor benignity.
Immunohistochemical studies show positivity for fibrillar glial acid protein (PGFA) and S100 in cells surrounding the pseudo papillae, as well as immunoreactivity for synaptophysin, neurofilaments, and OLIG2 in neuronal elements that are located in the interpapillary areas [9,10]. Tumor cells are negative for epithelial membrane antigen (EMA) and cytokeratins. The cell proliferation index measured with KI67 is generally low [7].
Molecularly, the fusion in the SLC4A1-PRCKCA gene has been found to be highly specific for this entity. Mutations in the BRAF V600E gene have also been described [2,11].
The treatment of these injuries is essentially surgical [1,2,12], associating this therapy modality with even injury-free progression and very good overall survival. In a meta-analysis [13] it is reported that the 1.5-year progression-free survival is 86% with an overall survival in the same period of 98%.
It has not been clarified in the reports that exist on this pathology if the clinical presentation is established as a risk factor for its greater aggressiveness, however, we are facing the first case of this entity that is associated with a paraneoplastic phenomenon of multiple multi topographic intracranial ischemic events that do not correlate with any other alteration other than the presence of this tumor entity. The paraneoplastic syndrome in the context of an oncological pathology at the level of the nervous system can have extensive involvement (brain, spinal cord, cranial nerves, retina, peripheral nerve and neuromuscular junction), being these alterations generally associated with tumor lesions with an expression of proteins with neuroendocrine potential, immunoglobulin production, or associated with organs with immune regulatory potential [14,15], characteristics that have not been described as present in papillary glioneural tumors and that open a spectrum of study about these apparently indolent tumors [5].
Conclusion
The papillary glial tumor, a rare entity and generally with a benign course, can show extremes of presentation, as discussed in this article. The need for greater knowledge about its natural history, immunobiology and treatment alternatives, opens a new window for the study of this pathology and establishes the need for a strict follow-up of patients who have this entity in order to learn more about its evolution. The first reported case associated with a presentation for neoplastic disease was described, which leads to question the "indolent" characteristic of this lesion and opens the question of a potential multi-systemic compromise associated with it.
References
- Carangel B, Arrigucci U (2015) Papillary glioneuronal tumor: Case report and review of literature. G Chir 36: 63-69.
- Goethe E (2019) Recurrent papillary glioneural tumor. World Neurosurg 128: 127-130.
- Komori T (1998) Papillary glioneuronal tumor: A new variant of mixed neuronal-glial neoplasm. Am J Surg Pathol 22: 1171-1183.
- Li D (2014) Clinical, radiological, and pathological features of 16 papillary glioneuronal tumors. Acta Neurochir (Wien) 156: 627-639.
- Adam C (2007) Papillary glioneural tumor: Not always a benign tumor? Clinical Neuropathology 26: 119-124.
- Doxtader E (2017) Cytopathologic features of papillary glioneuronal tumor. Diagnostic Cytopathology 1-3.
- Myung JK, Byeon SJ, Kim B, et al. (2011) Papillary glioneuronal tumors: A review of clinicopathologic and molecular genetic studies. Am J Surg Pathol 35: 1794-1805.
- Rocka S, Neverauskiene L, Nelson EL, et al. (2019) Papillary glioneuronal tumour: A case report. Cureus 11: e4215.
- Singh V, Gupta K, Salunke P, et al. (2019) Rosette-forming and papillary glioneuronal tumors - A clinicopathological and molecular analysis. Clin Neuropathol 38: 180-188.
- Matsumura N, Yokoo H, Mao Y, et al. (2013) Olig2-positive cells in glioneuronal tumors show both glial and neuronal characters: The implication of a common progenitor cell? Neuropathology 33: 246-255.
- Gelpi E (2007) Papillary glioneuronal tumor. Neuropathology 27: 468-473.
- Demetriades AK (2013) Papillary glioneuronal tumour: A review of the literature with two illustrative cases. Br J Neurosurg 27: 401-404.
- Schlamann A (2014) An individual patient data meta-analysis on characteristics and outcome of patients with papillary glioneuronal tumor, rosette glioneuronal tumor with neuropil-like islands and rosette forming glioneuronal tumor of the fourth ventricle PLoS One 9: e10121.
- Dalmau J (2008) Paraneoplastic syndromes of the CNS. Lancet Neurol 7: 327-340.
- Yadav N (2017) Papillary glioneuronal tumors: A radiopathologic correlation. Eur J Radiol 97: 44-52.
Corresponding Author
Alejandro Vargas-Moreno, Department of Neurosurgery, Hospital Universitario San Ignacio, Pontificia Universidad Javeriana, Cra. 7 #40-62, Sixth floor, Bogotá, Colombia, Tel: (+571)-5946161, Ext 2301.
Copyright
© 2021 Moreno AV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.