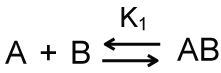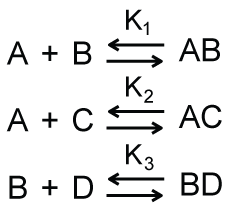Proteins are Not Recruited: A Plea for Better Diction
Abstract
Nearly all biological processes proceed or are controlled by protein-protein or protein-ligand binding reactions. Using anthropomorphic language to describe these interactions conveys an incorrect physical description of these processes while simultaneously minimizing the importance of the thermodynamics underpinning the associated interactions. Indeed, we should never say that proteins are recruited to binding partners or binding sites since this implies both a non-existent level of communication within biological systems and a non-existent process by which proteins or binding sites actively seek other proteins. Both of these fictions hinder our ability to determine quantitatively or qualitatively distinct biophysical descriptions of the associated systems. Here we present examples of how interactions typically described as protein recruitment can be more accurately and often more simply described as variations within binding equilibria. We argue that this approach is better for describing protein-protein and protein-ligand binding, even when the objective is only a qualitative description, especially for discussions with students in courses and research groups as it provides testable models for these interactions.
Introduction
It has become increasing common to say that proteins are recruited to their binding partners. For example, damage repair proteins are recruited to the sites of DNA damage [1,2] or the binding of one protein recruits the binding of additional proteins; examples of the latter include the formation of large DNA repair complexes [3], signal cascade networks [4-6], and even large scale and long range processes within cells [7]. While this terminology may be useful in expressing the idea that interactions exist between several macromolecules, it is nevertheless inappropriate since it simultaneously both implies a non-existent level of communication between biological macromolecules (i.e., how is a recruitment 'signal' propagated between macromolecules so that each becomes aware of the other's existence? Does this communication involve other macromolecules, as part of a cascade, e.g., as is often seen in other signaling networks?) and gives the false and anthropomorphic impression that biological macromolecules consciously seek out each other. Indeed, the word 'recruitment' implies an active process. Furthermore, this misleading characterization prevents a correct and thorough understanding of the thermodynamics (passively) underlying these interactions. While unintentional, using the terminology of recruitment can have negative pedagogical consequences since it implies that processes associated with recruitment are different from other interactions taught to the students such as cooperative binding, allosteric interactions, or a system of coupled binding equilibria. Using the term recruitment thus creates a distinction without a difference. Such a false distinction also imposes potential limitations on the creation of hypotheses for future experimental investigation of the associated interactions since it implies that recruitment is somehow different from changes in coupled thermodynamic equilibria (from allostery, cooperativity, e.g.). Since there as there is no mathematical model for recruitment-unless recruitment is used as a proxy for other well-characterized processes such as allosteric binding-the term is associated with no quantitative predictive power that can be tested experimentally. Lastly, even if the objective is only a qualitative rather than a quantitative description of a system of interactions, there is no advantage to use the terminology of recruitment rather than describing the system qualitatively in terms of allostery, cooperativity, coupled binding equilibria, etc., since the latter could be expanded into a quantitative description whereas the former cannot.
Here we advocate that interactions commonly referred to as protein recruitment instead be described as changes in apparent binding affinity resulting from perturbations of (potentially coupled) binding equilibria. We argue this language is better because it more accurately describes these interactions, allows for qualitative and quantitative predictions that can be especially investigated, and allows for the general fundamental principles governing them to be expressed more clearly. This encourages the pursuit of a common quantitative thermodynamic framework for characterizing these interactions, which in turn will facilitate the description of more complex processes-such as signaling or repair networks - that rely upon multiple interactions between proteins and ligands.
Simple 1:1 binding
Let's begin by considering the simple equilibrium binding reaction shown in Scheme 1. In this reaction, two macromolecules - A and B - bind to form a complex AB with 1:1 stoichiometry.
The variable K1 in Equation 1 is the equilibrium dissociation constant for the reaction. Equation 1 can be rearranged as an equation for the concentration of the complex.
The concentration of the complex thus depends upon the concentrations of the free macromolecules. These concentrations can be expressed in terms of the total concentrations of macromolecules, [Atotal] and [Btotal], using Equations 3 and 4.
Substitution of Equation 3 and Equation 4 into Equation 2 allows for the derivation of the commonly used quadratic equation for the concentration of the complex.
Equation 5 can be expressed more simply as Equation 6.
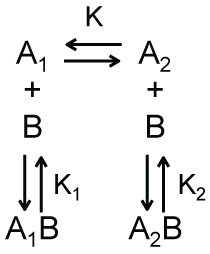
A trivial extension of this simple binding model is shown in Scheme 2a. In this case, there exists two different conformations for the macromolecule A, denoted as A1 and A2 in Scheme 2a, which have different affinities for binding the macromolecule B. The dimensionless equilibrium constant K, defined in Equation 7, relates the concentrations of A1 and A2.
As shown in Equation 7, a larger value of K corresponds to a larger concentration of A2 relative to A1. Following a derivation similar to that used to obtain Equation 6, we can obtain an expression for the total concentration of bound complexes.
The apparent dissociation constant Kapp in Equation 8 is shown in Equation 9.
Thus, when using an assay that is sensitive to only the presence of a complex, and not the individual species A1 and A2 within that complex, the measured equilibrium dissociation would be Kapp in Equation 9.
It follows from Equation 9 that the apparent equilibrium dissociation constant is a function of the equilibrium constant for two species of macromolecule A. As expected, when this equilibrium constant is large-i.e., when the equilibrium concentration of A2 exceeds that of A1-the apparent equilibrium dissociation constant approaches the equilibrium dissociation constant for the binding of A2 and B. Similarly, when this equilibrium constant is small-i.e., when the equilibrium concentration of A1 exceeds that of A2-the apparent equilibrium dissociation constant approaches the equilibrium dissociation constant for the binding of A1 and B.
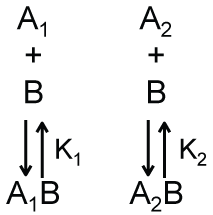
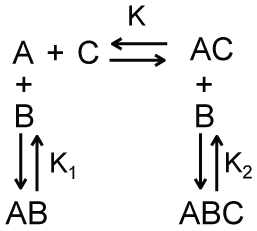
We argue that the model shown in Scheme 2b, which is a modified version of Scheme 2 in which there is no equilibrium between A1 and A2, can provide a simple description of the apparent recruitment of repair enzymes to a site of DNA damage. For this application, A1 represents a DNA binding site that is undamaged, A2 represents a DNA binding site that is damaged, and B represents a DNA damage repair protein. Let’s assume that the affinity of this repair protein to bind undamaged DNA is much weaker than its affinity to bind damaged DNA (i.e., K2 << K1). This difference in affinity may occur, for example, if the rate of dissociation is lower for interactions with damaged DNA than for interactions with undamaged DNA. Regardless, if this difference in binding affinity is large enough, repair proteins are more likely to be bound to damaged DNA rather than undamaged DNA even under conditions where the number of undamaged DNA binding sites far exceeds that of damaged DNA binding sites. This can be shown by solving the equations describing Scheme 2b for the ratio of the concentrations of the two bound complexes.
As shown in Equation 10, when K2 << K1, the concentration of repair proteins bound to damaged sites ([A2B]) can be much larger than the concentration of repair proteins bound to undamaged sites ([A1B]) even when the concentration of free undamaged sites ([A1]) is larger than the concentration of free damaged sites ([A2]). Thus, the apparent recruitment of repair proteins to locations of DNA damage occurs not because of some type of active communication between the DNA damage and the repair protein - as the work recruit implies-but rather as a result of the equilibrium thermodynamics describing the interactions between the repair protein and damaged and undamaged DNA. Furthermore, describing this process in terms of protein recruitment forgoes the opportunity and perhaps the motivation to understand the source of the variations in binding affinity within these binding equilibria. While one could make predictions about how single point mutations in a binding interface will affect binding affinity, how will such a mutation affect protein recruitment? This addresses the serious flaw that protein recruitment implies an active communication process by which proteins seek out one another. What is the mechanism of this communication? How do the proteins generate the associated signals and how do these signals propagate through the solution? Can these signals-perhaps transmitted as an additional macromolecule or cascade of macromolecules-be intercepted or otherwise hindered? We can find no answers to these questions in the literature.
Lastly, since Equation 10 provides a mathematical description of the binding interactions it enables quantitative examination of the associated processes such as their dependence on the total concentrations of the macromolecules or their dependence on temperature (through the temperature dependence of the equilibrium constants, e.g.). What is the prediction for how protein recruitment will depend upon temperature or free macromolecule concentration? If we observe that the equilibrium constant for a binding interaction displays a non-Arrhenius temperature dependence we would conclude a change in heat capacity occurred over the temperature range. How would one expect protein recruitment to change with temperature? Similarly, the equilibrium association constant will depend upon the diffusion coefficients of the macromolecules, which in turn will depend upon the viscosity of the solution (and temperature). What is the expectation for how protein recruitment will depend upon solution viscosity? Even if only a qualitative description is desired and thus the inability of a recruitment model to make quantitative predictions is immaterial, what is the advantage of qualitatively describing coupled binding equilibria or allostery as recruitment rather than as coupled binding equilibria or allostery?
There are several examples of DNA repair proteins that recognize specific structures of DNA [8-19]. Since the affinity of these proteins for binding these particular DNA structures is higher than their affinity for binding other DNA structures, these proteins would-according to the previously described mechanism-appear to be recruited to these particular types of DNA damage. However, as we have stated, a more appropriate description would be to say that the occurrence of DNA damage creates a specific binding site to which these proteins bind with particularly high affinity. As discussed above, the latter description/model has predictive power (what are the structures that bind with high affinity? What is the structural origin of binding selectivity for different types of damage by repair proteins? etc.) whereas the former does not. A similar mechanism is suggested for the early steps in prokaryotic gene expression. Specifically, the affinity of DNA binding by the TATA binding protein depends upon the sequence of the DNA, being highest for prompter sequences containing the TATA box [20]. This variation in affinity allows this protein to find promoter sequences through a process of facilitated diffusion along the DNA [21]. Namely, the ability of the TATA binding protein to bind nonspecifically to DNA enables the protein to translocate or slide along the DNA until it locates (and binds more tightly to) the prompter sequence [21]. A similar mechanism is argued to govern the E. coli lac repressor-operator interaction [22-31]. All of these processes can be described qualitatively or quantitatively without ever using the language of recruitment and, perhaps more importantly, the use of recruitment terminology has no advantage over describing these processes in terms of coupled binding equilibria.
Sequential binding
There are many examples of sequential binding events in molecular biology in which the binding of two macromolecules allosterically regulates the binding of a third macromolecule. For example, in eukaryotic transcription it is commonly stated that the binding of transcription factors to DNA recruits RNA polymerase II [32-40]. Rather, we should instead describe such sequential binding interactions using coupled binding equilibria.
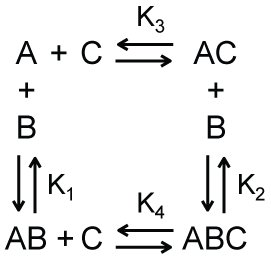
A simple model for sequential binding interactions is shown in Scheme 3. In this model, macromolecule A binds macromolecule B with an equilibrium dissociation constant K1 and macromolecule C with an equilibrium dissociation constant K. When in complex with macromolecule C, macromolecule A can still bind macromolecule B, but now with equilibrium dissociation constant K2. Following a derivation similar to that used to obtain Equation 6, we can obtain an expression for the total concentration of bound complexes.
The apparent equilibrium dissociation constant Kapp in Equation 11 is shown in Equation 12.
As shown in Equation 12, the apparent equilibrium dissociation constant is a function of both the equilibrium dissociation constant for the binding of macromolecule A and macromolecule C, and the concentration of free macromolecule C. As expected from Scheme 3, an increase in either of these quantities shifts the apparent equilibrium dissociation constant toward that describing the binding of the complex AC with macromolecule B.
Returning to the example of eukaryotic gene expression, we could denote the transcription factor as macromolecule C, and K1 and K2 as the equilibrium dissociation constants for the binding of RNA polymerase II to the DNA in the presence and absence, respectively, of the transcription factor. Let's then further assume that K2 < K1, so that the complex AC binds macromolecule B with higher affinity that macromolecule A binds macromolecule B. As indicated by this model and shown in Equation 12, the presence (i.e., binding of) the transcription factor allosterically shifts the thermodynamic equilibrium of DNA binding by RNA polymerase II to a higher affinity reaction. To put it differently, there is cooperativity within the equilibrium binding of the transcription factor, RNA polymerase II, and DNA.
Indeed, we could define a cooperativity parameter w to describe the allosteric change in the affinity of macromolecule A binding to macromolecule B in the presence of macromolecule C. Let's define the relationship between w, K1, and K2 using Equation 13.
Substitution of Equation 13 into Equation 12 thus yields
Thus, positive cooperativity (w > 1) would result in behavior typically described as recruitment; specifically, the binding of macromolecule C increases the affinity of the interaction between macromolecule A and macromolecule B. Negative cooperativity (0 < w < 1) would result in the opposite of recruitment; specifically, the binding of macromolecule C decreases the affinity of the interaction between macromolecule A and macromolecule B. It is not immediately clear, however, how negative cooperativity can be anthropomorphized.
For the model shown in Scheme 3, we assumed that there is a difference in the affinity of the interaction between macromolecule A and macromolecule B when macromolecule C is bound to macromolecule A. In other words, we assumed that K1 and K2 are different (or that the cooperativity parameter w is not equal to one). This may result, for example, because the binding of macromolecule C changes the structure of macromolecule A and thus the binding interface and affinity for macromolecule B. Alternatively, it may be the case that the binding of macromolecule C provides an additional interface for the binding of macromolecule B. In this case, macromolecule B is binding to both macromolecule A and macromolecule C simultaneously. This is argued to occur, for example, during the formation of the eukaryotic transcription complex [34]. Of course, these two outcomes are not mutually exclusive and both combined can affect the binding of macromolecule B. Describing these interactions using cooperativity and coupled binding equilibria prompts (and allows for) quantitative predictions that can be empirically tested; this is not possible with a recruitment model. The former descriptions also motivate investigations into the origin of the allostery and/or the extent of the coupled binding equilibria. Are there structural changes that give rise to these effects and if so what are they? The terminology of recruitment prompts no such questions and allows for no additional hypothesis since it is associated with no underlying testable model.
Let us explore this point further by considering the canonical example of allostery-oxygen binding by hemoglobin. If we described this system by saying the binding of oxygen molecules by hemoglobin recruited subsequent additional oxygen molecules to hemoglobin, how would we explain why the presence of carbon dioxide, protons, and 2,3-Bisphosphoglycerate affect oxygen recruitment? How does the presence of these molecules affect the communication between hemoglobin and oxygen that the terminology of recruitment implies? Furthermore, could a recruitment model provide an equation to fit a plot of the fractional saturation of oxygen binding to hemoglobin as a function of the partial pressure of oxygen or the dependence of these plots on solution pH? Since recruitment has no qualitative or quantitative predictive power it has no advantage in describing this system, which likely accounts for why we can find no recruitment-based description of it in the literature.
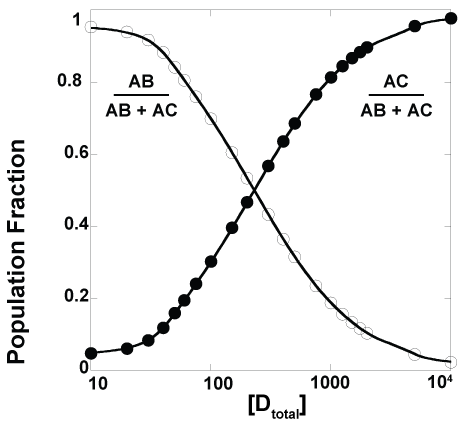
It is, of course, possible to have a system such as that shown in Scheme 4 in which one macromolecule, denoted as A, has one binding site which can be occupied by one of two possible ligands, denoted as B and C. The B ligand can also bind a different macromolecule, denoted as D. As shown in Figure 1, the relative faction of the AB and AC complexes is a function of the total concentration of macromolecule D; this dependence is, ofcourse, also a function of the equilibrium dissociation constants and the total concentrations of the other macromolecules. For the simulations shown in Figure 1, the equilibrium dissociation constants K1, K2, and K3 in Scheme 4 were identical. Unsurprisingly, similar traces are obtained when these equilibrium constants are different from one another or when different total concentrations of macromolecules A, B, and C are used.
A frequently encountered example of a system that can be described approximately by Scheme 4 is the use of the lac promoter to control gene expression. The binding of lac repressor prevents the binding of RNA polymerase to the promoter and thus these two complexes exhibit what can be described as complete negative cooperativity for binding to the promoter. IPTG competes with the lac promoter for binding lac repressor and thus in the presence of IPTG the binding equilibria will shift toward favoring lac repressor bound to IPTG rather than to the lac promoter. This will leave the lac promoter available to binding by RNA polymerase. This accounts for the ability of IPTG to trigger expression of lac promoter controlled genes and why leaky expression occurs in these systems. Indeed, there will always be a small fraction of lac promoters bound by RNA polymerase, even under conditions of excess lac repressor, and thus a small level of gene expression (Figure 1). Significant for this conversation is the fact that this behavior can be described effectively and succinctly through this model of coupled equilibria with no need of protein recruitment. Indeed, one does not say that the removal of lac repressor recruits the binding of RNA polymerase to the lac promoter nor that the lac repressor recruits IPTG.
Finally, let's consider the system shown in Scheme 5 in which macromolecule A has two different binding sites for two different ligands, denoted as B and C in Scheme 5. An example of such a system of binding is found within translocases, which must bind both nucleotide (ATP, ADP, e.g.) and their translocation substrate (DNA, RNA, polypeptide, e.g.) [41-48]. It is also common that the affinities of these motors for binding nucleotide and binding the translocation substrate are allosterically linked to each other [49-51]. In fact, the regulation of substrate binding affinity by differences in the affinity of binding ATP and ADP forms the basis of some models for processive translocation by these motors [51,52]. Specifically, variations in the affinity of substrate binding within the ATPase cycle (associated with ATP, ADP, or no nucleotide bound by the motor) allows the motor to dissociate from and rebind to the translocation substrate in a periodic manner that gives rise to translocation. What is absent from studies of these systems, however, are statements that the binding of ATP or ADP recruits the binding of the translocation substrate, or vice versa. Instead, these systems are always described in terms of coupled thermodynamic equilibria, such as those shown in Scheme 5. We advocate that this example be followed more widely in studies of protein-protein and protein-ligand interactions.
Conclusion
The words we choose to describe science affect not only the ability of our audience to understand it, but also our deeper understanding of it. For example, entropy is too often wrongly described as a measure of disorder. Thinking about entropy as a measure of disorder prevents an accurate understanding of how to use entropy to calculate the thermodynamic properties of a system (heat capacity, pressure, etc.) and by extension how to incorporate entropy correctly into a thermodynamic description of a system. Even if one is seeking only a qualitative description of entropy or its effects, there is no benefit to using an incorrect description that invokes disorder.
Similarly, describing protein-protein or protein-ligand interactions in terms of recruitment gives the false impression to our students, our peers, and ourselves that an underlying active communication network exists between biological macromolecules and that the process or processes associated with protein recruitment are distinct from cooperative binding, an allosteric interaction, or a system of coupled binding equilibria. This further implies that the thermodynamics associated with recruitment are different from those associated with these processes [53]. This is simply not true and causes confusion. It would be far better to embrace quantitative thermodynamic and by extension statistical mechanical descriptions of macromolecular interactions, especially given the central role of these interactions to nearly all biological processes.
Furthermore, by actively ignoring the coupled binding equilibria existing within these systems we fail to motivate our students, our peers, and ourselves to understand and appreciate the thermodynamics that truly governs these processes. The associated resulting misconceptions hinder progress since they hamper the development of physically appropriate questions for future experiments. Indeed, since there is no physical or mathematical model for recruitment, a recruitment model can make no qualitative or quantitative predictions that can be tested empirically. As mentioned above, how would one expect protein recruitment to vary with solution conditions (temperature, ionic strength, etc.) or with the concentrations of the macromolecules involved in the interaction (or with the concentrations of other macromolecules in the solution)? And even if only a qualitative description of a network of interactions is the goal, using the terminology of recruitment offers no advantage over descriptions involving cooperativity, allostery, or coupled equilibria. Since there is no benefit to using a recruitment model to describe macromolecular interactions and several associated disadvantages, we advocate that the terminology of recruitment never be used.
References
- Hable V, Drexler GA, Brüning T, et al. (2012) Recruitment Kinetics of DNA Repair Proteins Mdc1 and Rad52 but Not 53BP1 Depend on Damage Complexity. PLoS One 7: e41943.
- Hong Z, Jiang J, Hashiguchi K, et al. (2008) Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J Cell Sci 121: 3146-3154.
- Ding N, Bonham EM, Hannon BE, et al. (2016) Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J Mol Cell Biol 8: 244-254.
- Ono YJ, Tanabe A, Tanaka T, et al. (2015) Met Signaling Cascade Is Amplified by the Recruitment of Phosphorylated Met to Lipid Rafts via CD24 and Leads to Drug Resistance in Endometrial Cancer Cell Lines. Mol Cancer Ther 14: 2353-2363.
- Nelson JD, LeBoeuf RC, Bomsztyk K (2011) Direct Recruitment of Insulin Receptor and ERK Signaling Cascade to Insulin-Inducible Gene Loci. Diabetes 60: 127-137.
- Pryciak PM, Huntress FA (1998) Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gbeta gamma complex underlies activation of the yeast pheromone response pathway. Genes Dev 12: 2684-2697.
- Finlan LE, Sproul D, Thomson I, et al. (2008) Recruitment to the Nuclear Periphery Can Alter Expression of Genes in Human Cells. PLoS Genet 4: e 1000039.
- Wang L, Lee SJ, Verdine GL (2015) Structural Basis for Avoidance of Promutagenic DNA Repair by MutY Adenine DNA Glycosylase. J Biol Chem 290: 17096-17105.
- Banerjee A, Yang W, Karplus M, et al. (2005) Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 434: 612-618.
- Labahn J, Schärer OD, Long A, et al. (1996) Structural Basis for the Excision Repair of Alkylation-damaged DNA. Cell 86: 321-329.
- Wang Y, Cortez D, Yazdi P, et al. (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927-939.
- Fadda E (2016) Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput Struct Biotechnol J 14: 78-85.
- Keyamura K, Sakaguchi C, Kubota Y, et al. (2013) RecA protein recruits structural maintenance of chromosomes (SMC)-like RecN protein to DNA double-strand breaks. J Biol Chem 288: 29229-29237. .
- Ogi T, Limsirichaikul S, Overmeer RM, et al. (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell 37: 714-727.
- Sugasawa K (2011) Multiple DNA damage recognition factors involved in mammalian nucleotide excision repair. Biochemistry 76: 16-23.
- Sugasawa K (2016) Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair. DNA Repair (Amst) 44: 110-117.
- Tsodikov OV, Ivanov D, Orelli B, et al. (2007) Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J 26: 4768-4776.
- Volker M, Moné MJ, Karmakar P, et al. (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 8: 213-224.
- Wong I, Bernards AS, Miller JK, et al. (2003) A dimeric mechanism for contextual target recognition by MutY glycosylase. J Biol Chem 278: 2411-2418.
- Weideman CA, Netter RC, Benjamin LR, et al. (1997) Dynamic interplay of TFIIA, TBP and TATA DNA. J Mol Biol 271: 61-75.
- Coleman RA, Pugh BF (1995) Evidence for functional binding and stable sliding of the TATA binding protein on nonspecific DNA. J Biol Chem 270: 13850-13859.
- Berg OG, Winter RB, von Hippel PH (1981) Diffusion-driven mechanisms of protein translocation on nucleic acids 1 Models and theory. Biochemistry 20: 6929-6948.
- Winter RB, von Hippel PH (1981) Diffusion-driven mechanisms of protein translocation on nucleic acids 2 The Escherichia coli repressor-operator interaction: equilibrium measurements. Biochemistry 20: 6948-6960.
- Winter RB, Berg OG, Von Hippel PH (1981) Diffusion-Driven Mechanisms of Protein Translocation on Nucleic Acids. 3. The Escherichia coli lac Repressor-Operator Interaction: Kinetic Measurements and Conclusions. Biochemistry 20: 6961-6977.
- Hsieh WT, Wells RD (1987) Influence of negative supercoiling and of the proximity of left-handed Z-DNA on the Escherichia coli lactose repressor-operator interaction. J Biol Chem 262: 14576-14582.
- Barkley MD, Riggs AD, Jobe A, et al. (1975) Interaction of effecting ligands with lac repressor and repressor-operator complex. Biochemistry 14: 1700-1712.
- Gatti-Lafranconi P, Dijkman WP, Devenish SR, et al. (2013) A single mutation in the core domain of the lac repressor reduces leakiness. Microb Cell Fact 12: 67.
- Paquet F, Maurizot JC, Lancelot G (2000) Changes in lac Operator dynamics upon selective interaction with lac Repressor as revealed by heteronuclear relaxation rate measurements. Magn Reson Chem 38: 946-950.
- O'Gorman RB, Rosenberg JM, Kallai OB, et al. (1980) Equilibrium binding of inducer to lac repressor operator DNA complex. J Biol Chem 255: 10107-10114.
- Wilson CJ, Zhan H, Swint-Kruse L, et al. (2007) The lactose repressor system: paradigms for regulation, allosteric behavior and protein folding. Cell Mol Life Sci 64: 3-16.
- Zuo Z, Chang Y, Stormo GD (2015) A quantitative understanding of lac repressor's binding specificity and flexibility. Quant Biol 3: 69-80.
- Miesfeld R, McEvoy M (2017) Biochemistry. (1st edn), First W W Norton & Company.
- Berg JM, Tymoczko JL, Stryer L (2006) Biochemistry.
- Cooper GM (2000) Eukaryotic RNA Polymerases and General Transcription Factors. In: The Cell: A Molecular Approach. (2nd edn), Sinauer Associates Inc.
- Lodish H, Berk A, Zipursky SL, et al. (2000) RNA Polymerase II Transcription-Initiation Complex. In: Molecular Cell Biology. (4th edn), WH Freeman & Co Ltd.
- Barberis A, Müller CW, Harrison SC, et al. (1993) Delineation of two functional regions of transcription factor TFIIB. Proc Natl Acad Sci U S A 90: 5628-5632.
- Flores O, Lu H, Killeen M, et al. (1991) The small subunit of transcription factor IIF recruits RNA polymerase II into the preinitiation complex. Proc Natl Acad Sci U S A 88: 9999-10003.
- Ha I, Roberts S, Maldonado E, et al. (1993) Multiple functional domains of human transcription factor IIB: distinct interactions with two general transcription factors and RNA polymerase II. Genes Dev 7: 1021-1032.
- Sainsbury S, Bernecky C, Cramer P (2015) Structural basis of transcription initiation by RNA polymerase II. Nat Rev Mol Cell Biol 16: 129-143.
- Zhao X, Herr W (2002) A regulated two-step mechanism of TBP binding to DNA: a solvent-exposed surface of TBP inhibits TATA box recognition. Cell 108: 615-627.
- Pyle AM (2008) Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37: 317-336.
- Smita S Patel, Ilker Donmez (2006) Mechanisms of helicases. J Biol Chem 281: 18265-18268.
- Lohman TM, Tomko EJ, Wu CG (2008) Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat Rev Mol Cell Biol 9: 391-401.
- Becker PB (2005) Nucleosome remodelers on track. Nat Struct Mol Biol 12: 732-733.
- Becker PB, Hörz W (2002) ATP-dependent nucleosome remodeling. Annu Rev Biochem 71: 247-273.
- Feiss M, Rao VB (2012) The bacteriophage DNA packaging machine. Adv Exp Med Biol 726: 489-509.
- Stanley LK, Seidel R, van der Scheer C, et al. (2006) When a helicase is not a helicase: dsDNA tracking by the motor protein EcoR124I. EMBO J 25: 2230-2239.
- Smith RM, Josephsen J, Szczelkun MD (2009) The single polypeptide restriction-modification enzyme LlaGI is a self-contained molecular motor that translocates DNA loops. Nucleic Acids Res 37: 7219-7230.
- Wong I, Lohman TM (1992) Allosteric effects of nucleotide cofactors on Escherichia coli Rep helicase-DNA binding. Science 256: 350-355.
- Malik SS, Rich E, Viswanathan R, et al. (2011) Allosteric interactions of DNA and nucleotides with S. cerevisiae RSC. Biochemistry 50: 7881-7890.
- Timothy M Lohman, John Hsieh, Nasib K Maluf, et al. (2003) DNA helicases, motors that move along nucleic acids: Lessons from the SF1 helicase superfamily. The Enzymes 23: 303-369.
- Briggs K, Fischer CJ (2014) All motors have to decide is what to do with the DNA that is given them. Biomol Concepts 5: 383-395.
- Lewis M (2011) A tale of two repressors. J Mol Biol 409: 14-27.
Corresponding Author
Christopher J Fischer, Department of Physics and Astronomy, The University of Kansas, 1251 Wescoe Hall Drive, 1082 Malott, Lawrence, KS 66045-7582, USA, Tel: 785-864-4579, Fax: 785-864-5262.
Copyright
© 2017 Grotemeyer EN, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.