Mycobacterium Tuberculosis Drug Resistance Mechanisms
Abstract
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (MTB). It affects mainly the lungs, but it may also occur in extrapulmonary sites. TB is a world challenge due to its high burden and the emergency of drug-resistant MTB. MTB employs a wide range of resistance mechanisms to escape the action of the drugs currently in use for TB treatment. These mechanisms can be classified as intrinsic or acquired and are related to mycobacterium genetic diversity, strain transmission, and irregular treatment. The surveillance of resistant MTB is of high importance to guide treatment recommendations and control measures.
Keywords
Mycobacterium tuberculosis , Drug sensitivity, Mutation, Genomic surveillance
Abbreviations
TB: Tuberculosis; MTB: Mycobacterium tuberculosis ; WHO: World Health Organization; H: Isoniazid; R: Rifampin; Z: Pirazinamide; E: Ethambutol; DR-TB: Drug-resistant TB; MDR-TB: Multidrug-resistant TB; AIDS: Acquired Immunodeficiency Syndrome; LPA: Line Probe Assay; PCR: Polymerase-Chain-Reaction; WGS: Whole Genome Sequencing; RR-TB: Rifampin-Resistant TB; FQ: Fluoroquinolone; XDR-TB: Extensively Drug-Resistant TB; LVX: Levofloxacin; MOX: Moxifloxacin; BDQ: Bedaquiline; LZD: Linezolid; ETA: Ethionamide; CFZ: Clofazimine; SITs: Spolygotype International Types
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), is among the worldwide deadliest infectious diseases caused by a single agent [1]. This bacillus is transmitted mainly via aerosol droplets expelled by individuals with active disease. TB affects primarily the lungs but is also responsible for extrapulmonary infections. The majority of infected individuals will progress to a latent phase of the disease, in which the quiescent bacillus does not elicit the classic signs of TB and is not transmissible to novel hosts. Active TB develops in 5-10% of this pool of infected individuals (secondary or post-primary TB), especially among immune compromised subjects [2].
Robert Koch isolated the bacillus in 1882, and anti-TB treatment was initiated in 1944, as the first drug active against MTB, streptomycin (S) was characterized [3]. Isoniazid (H) was introduced in 1954 and constituted a milestone in TB treatment, given its efficacy against the bacillus, and the fact that it was the first orally administered drug developed. Rifamycin B, the precursor drug of rifampin (R), was introduced in TB treatment in 1964 [4,5].
Mycobacterial resistance against the drugs employed in TB treatment occurred in parallel with drug discovery and adoption in anti-TB regimens. The first national study of resistance against anti-TB drugs was conducted in Great Britain in the mid-1950s [6]. By the end of the 1990s, the rise in TB cases associated with the AIDS pandemic and the surge of TB drug-resistant strains motivated the intensification of TB control strategies [7]. TB was considered a global emergency by the World Health Organization (WHO) in 1993 [8].
WHO currently recommends the use of four drugs for the first-line treatment against drug-sensitive TB: R, H, pyrazinamide (Z), and ethambutol (E) used in a two-phase regimen, in which all four drugs are administered for 2 months in the first (attack) phase and R plus H is maintained for additional 4 months in the second (maintenance) phase. However, in case of therapeutic failure and/or resistance detection, the treatment must be adjusted according to the resistance profile [9].
Globally, it is estimated that some degree of drug resistance occurs in one out of three patients [1]. India, China, and the Russian Federation host more that 50% of global drug-resistant TB (DR-TB) cases worldwide [10]. The combined resistance to R and H is of particular concern, as these are the two most effective drugs of the first-line regimen, and resistance against them is associated with increased rates of unfavorable outcomes. The seven countries with the highest TB incidence are responsible for approximately 2/3 of reported cases with this combined drug resistance, termed multidrug-resistant TB (MDR-TB) [1]. DR-TB is a severe public health problem with a global impact, especially among developing countries. Many factors may influence the emergence of drug-resistant MTB strains, such as bacterial fitness, environmental factors, and host-related factors including genetic background [11].
Drug sensitivity tests can be performed to detect resistance phenotypically, from cultured isolates [12]. Testing is usually performed in liquid cultures. This is considered a gold-standard test to inform the therapeutic management of TB patients [13]. The limitations of phenotypic tests include the long period necessary to obtain the results, the laboratory infrastructure needed to cultivate the bacillus following the biosafety norms applicable, and the challenges posed by drug dose effect and intra-host variability in drug sensitivity [13].
Molecular tests detect mutations in genes associated with drug resistance and are increasingly used as alternatives to phenotypic testing. Some of the broadly-used molecular tests to determine drug resistance constitute variations of the line probe assay (LPA), which are based on the amplification of segments of the MTB genome (e.g. the gene rpoB , involved in resistance to R) by polymerase-chain-reaction (PCR). Mutated sequences are recognized by hybridization with specific probes linked to detection molecules. Other widely employed tests are based on real-time quantitative PCR, and use molecular beacon technology to determine the Mycobacterial species and resistance to R as a screening strategy to detect MDR-TB, as well as resistance to other drugs [14-16].
Initially used in the context of research and surveillance, to monitor the resistance profiles of MTB, whole genome sequencing (WGS) is proving to be a reliable, fast and increasingly affordable tool to be used for patient management [17]. WGS allows for the simultaneous characterization of multiple genome sites involved in drug resistance; furthermore, it makes possible to identify new mutations that may confer phenotypic resistance to anti-TB drugs. It also allows for the identification of mixed infections and of the MTB strain(s) recovered from the patient, in addition to enabling individualized treatment [17]. The WHO points to the WGS as a useful tool in identifying transmission chains, outbreaks and the emergence of resistance to current and new drugs that can be used against TB [12].
Drug resistance impacts TB control and constitutes a threat to disease eradication. Developing countries face difficulties in broadening the coverage of drug-sensitivity tests, which impacts disease burden, patient management, and DR-TB surveillance. WHO emphasizes the need to monitor at least 80% of new bacteriologically confirmed TB pulmonary cases and previously treated TB cases for RR-TB (Rifampin-resistant TB). Among RR-TB cases, the WHO urges the identification of fluoroquinolone (FQ) resistance [12].
Resistance to Single and Combined Anti-TB Drugs
Drug-resistant TB is defined as the disease caused by MTB resistant to one or more drugs used in TB treatment in laboratory tests [18]. DR-TB cases have increased since the first drug, streptomycin (S), was approved for use, in 1948 [19]. Presently DR-TB is a severe public health problem and the major contributor to worldwide antimicrobial resistance. Approximately 0.5 million persons become infected with DR-TB annually [18].
Resistant MTB strains usually evolve by mutations in genes that encode the pharmacological targets of anti-TB drugs, or in those that encode enzymes involved in the metabolization of pro-drugs into active forms [20,21]. Mobile genetic elements characteristic of other bacteria are not found in MTB and therefore do not contribute to Mycobacterial resistance [21].
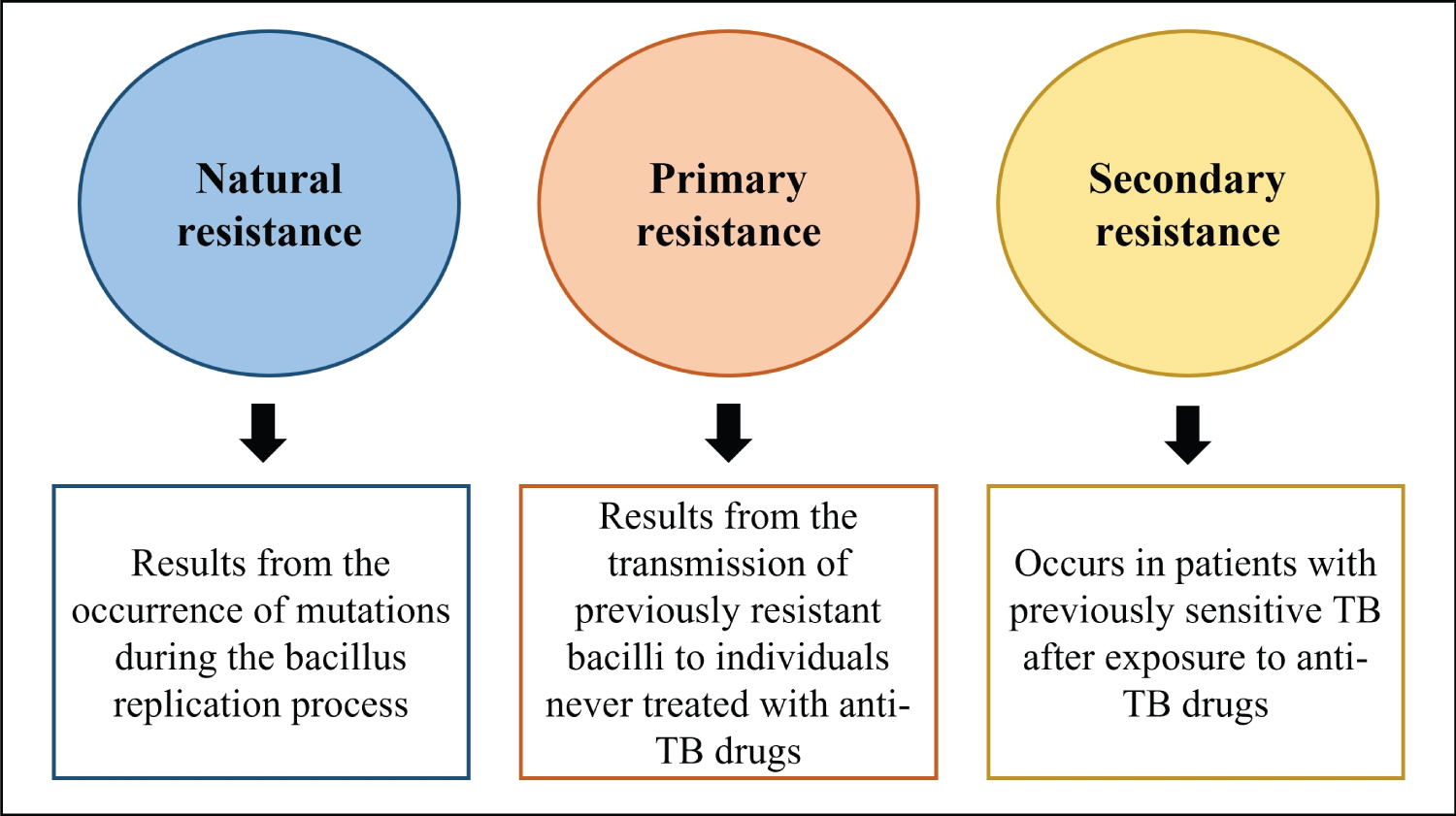
Natural drug resistance may result from bacilli multiplication, involving aleatory point mutations [21]. However, treatment imposes a selection pressure that can favor resistant strains in the host, and irregular drug regimens e.g. due to low adherence are associated with DR-TB [22]. Individuals that have never received anti-TB treatment may also present resistant bacilli when infected with MTB from DR-TB patients. Disease resulting from host infection with drug-resistant MTB strains is termed primary resistant TB. DR-TB that evolves in a patient due to drug selection pressure is termed secondary resistant TB [23]. Figure 1 summarizes the classification of TB resistance.
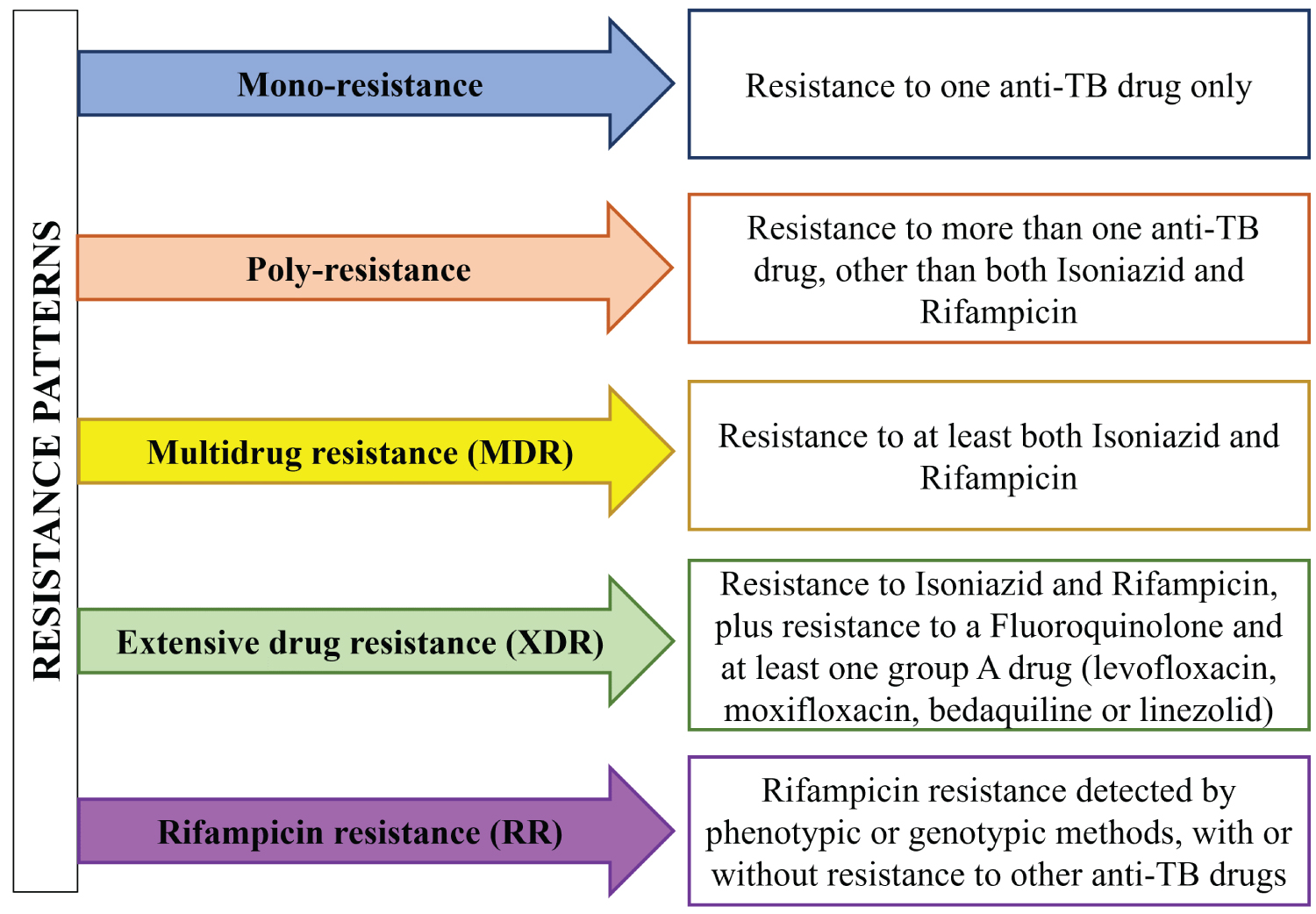
WHO classifies DR-TB into five types, considering which drugs are involved and single vs combined resistance to two or more drugs (Figure 2). The term mono resistant TB corresponds to resistance to a single first-line drug. Combined resistance to two or more drugs is classified as poly-resistant TB, except for the combined resistance to R and H, termed multidrug-resistant (MDR-TB). MDR-TB can be further characterized as pre-XDR-TB (Extensively drug-resistant TB), in patients with resistance to R, H, and any FQ, and XDR-TB, in patients with resistance to R, H, any FQ and at least one group A drug, which presently comprises the drugs levofloxacin (LVX), moxifloxacin (MOX), bedaquiline (BDQ) and linezolid (LZD) [12]. RR-TB corresponds to any form of TB resistant to R, detected by genotypic or phenotypic methods, with or without further characterization of the drug susceptibility to other anti-TB drugs [18]. XDR-TB is the rarest type of DR-TB, associated with limited treatment options, which are generally more toxic and less effective, and require a longer administration time [24].
The World Health Organization estimates that in 2021 there was a reversal in the stabilization of the number of cases of MDR-TB and RR-TB observed between 2015 and 2020. This year, the data show a growth of 3.1% in the number of estimated cases compared to the previous year, totaling 450,000 new cases worldwide. Thus, MDR-TB or RR-TB cases represented 3.6% of all TB cases, and 18% of the previously treated TB cases, in 2021 [1].
WHO presently proposes new treatment categories of shorter duration for MDR/RR-TB [25,26]. The 6-month regimen combines the administration of bedaquiline, pretomanid, linezolid, and moxifloxacin (BPaLM), and the 9-month regimen combines only orally administered drugs (BDQ, LVX or MOX, ethionamide - ETA or LZD, E, high-dose H, Z and clofazimine - CFZ). The former is recommended for MDR/RR-TB with or without resistance to FQ, but BPaLM should be substituted for BPaL (with the discontinuation of MOX), while the 9-month oral regimen should not be administered to individuals with FQ-resistant TB [26]. The long-term 18-month regimen is recommended in cases where shorter regimens are contraindicated [26]. It combines drugs from group A (LVX/MOX, BDQ, and LZD) with at least one drug from group B (CFZ and cycloserine/terizidone), ensuring the use of 4 different drugs. If the treatment cannot consist only of drugs from groups A and B, those from group C (E, delamanid, Z, imipenem-cilastatin/meropenem, amikacin/streptomycin, ETA, or prothionamide and P-aminosalicylic acid) can be used. Treatment must be guided by the drug-susceptibility profiling of the patients [25].
Genetic Diversity of MTB and Resistance to Anti-TB Drugs
The MTB complex comprises 7 lineages of Mycobacterial that are strictly adapted to cause a complete cycle of infection only in humans. These lineages are thought to have evolved in parallel with humans, accompanying the human diaspora from the African continent, which would explain the geographic association between MTB lineages and human populations [27,28] Lineages 1-4 and lineage 7 correspond to M. tuberculosis , while lineages 5 and 6 correspond to M. africanum [29]. They can also be classified in families, subfamilies and spolygotype international types (SITs) by classic genetic approaches. MTB spoligotype families include Beijing, Central Asia Strain (CAS), East African Indian (EAI), Haarlem (H), Latin-American Mediterranean (LAM), T, and X.
MTB lineages differ in transmission, mutation rates, biological fitness, virulence, and in the immune responses they elicit in the host [30]. Highly transmitted strains are also more likely to acquire drug resistance mutations, although the exact relation between drug resistance accumulation and strain genetic background is poorly understood [31,32]. The CRyPTIC Consortium mapped the genomes of over 12 thousand MTB global clinical isolates to associate their genetic profiles to their phenotypic resistance against 13 anti-TB drugs.
MTB Intrinsic Resistance Mechanisms
Cell wall composition
The intrinsic resistance of mycobacterium against various classes of antibiotics has commonly been attributed to the special composition and structure of the Mycobacterial cell wall [33]. Unlike other Gram-positive bacteria, the Mycobacterial wall is thick and contains a wide range of different lipids, including mycolic acids [34,35]. Its lipid-rich nature makes this wall extremely hydrophobic, preventing the permeation of hydrophilic compounds, including several antibiotics belonging to the macrolide, Rifamycin, tetracycline, and FQ classes. The low number of porins significantly contributes to this reduced permeability [35,36].
Mutations in several genes associated with cell wall biosynthesis are related to resistance to anti-TB drugs: kas A, ahp C, ni A, ndh , fadE24 , fabG1 , embB , and embC [37-39]. Additionally, the heterologous expression of porin MspA in MTB decreased the minimum inhibitory concentration for several hydrophilic drugs, indicating that porins play an important role in the diffusion of hydrophilic antibiotics via the cell wall [40,41]. The hypothesis that cell wall lipids are an important factor in the intrinsic resistance of Mycobacterial to many hydrophobic antibiotics is supported by studies carried out with defective mutants in lipid synthesis. These mutants are susceptible to drugs, unlike the resistant wild-type strain [42].
Drug inactivation
After penetrating via the cell wall, antibiotics can be enzymatic ally cleaved in the periplasmic space and become ineffective. One example of inactivation is the enzymatic degradation of β-lactam antibiotics by β-lactamases, which hydrolyze the β-lactam ring. The first studies involving penicillin demonstrated that MTB is intrinsically resistant to this class of antibiotics [43]. The MTB genome encodes BlaC (Rv2068c), a broad-spectrum and highly active β-lactamase [44]. This β-lactamase has broad substrate specificity, including carbapenems, fulfilling the definition of extended spectrum. BlaC is irreversibly inhibited by clavulanate [45,46]. Therefore, the combined treatment using carbapenem, a β-lactam capable of inhibiting Ldts and D,D-carboxypeptidases from Mycobacterial, with a β-lactamase inhibitor, clavulanic acid, which inhibits BlaC, was included in the anti-TB drug repertoire [47-49]. However, point mutations may occur in BlaC, allowing for the hydrolysis of clavulanic acid [50-52]. Accordingly, some XDR-TB isolates were shown to be insensitive to mereponem/clavulanate or amoxicillin/clavulanate treatment, even without harboring any mutations linked to the susceptibility to these drugs. Nevertheless, the safety profile of β-lactam antibiotics/β-lactamase inhibitors and the limited treatment options for MDR/XDR-TB warrants further investigation into new treatment regimens including this class of antibiotics.
In addition to drug cleavage, antibiotics can be inactivated by modifications such as methylation or acetylation. The pyrido-benzimidazole compound 14 has potent bactericidal activity against aerobically growing Mtb [53]. This compound can be methylated by a methyltransferase encoded by the constitutive gene Rv0560c in Mtb. The methylated compound 14 is unable to inhibit its target, decaprenylphosphoryl-β-D-ribose 2-oxidase (DprE1), a molecule involved in the synthesis of arabinogalactan [54]. However, this new mechanism of drug resistance in Mtb has no known clinical relevance so far. On the other hand, the intracellular survival protein (Eis)-mediated acetylation of amino glycoside/cyclic peptide antibiotics used to treat MDR-TB is a mechanism leading to drug inactivation. Eis acetylates and inactivates second-line injectable amino glycosides such as kanamycin A, as well as cyclic peptides such as capreomycin [55-57]. Mutations leading to Eis over expression were identified in clinical isolates of MTB, which showed resistance to kanamycin A [57,58]. Eis over expression, therefore, may foster the evolution of high-level resistance to amino glycosides and cyclic peptides.
Efflux pumps
Efflux pumps are protein transport channels that pump various compounds, including antibiotics, out of the cell. The MTB efflux pumps are located in the plasma membrane, which is part of a complex cell envelope that is responsible for protecting the bacteria in hostile environments, mechanical resistance, transport of solutes and proteins, and adhesion to host receptors, in addition to having the important role of intrinsic antimicrobial resistance [59]. Several families of efflux pumps in MTB and other Mycobacterial have been characterized as antibiotic carriers [60]. Five super families encoding known or putative transporters are found in MTB: the ATP-binding cassette (ABC), the Major Facilitator Super-family (MFS), the Resistance Nodulation Division (RND), the Small Multidrug Resistance (SMR), and the Multidrug and Toxic-Compound Extrusion (MATE) [59].
While specific mutations in efflux-associated genes have been linked to resistance to several anti-TB drugs, up regulation of efflux pump expression also functions as an adaptive mechanism that can be activated by drug pressure in genetically sensitive MTB strains [42]. The expression of efflux pumps is condition-dependent, reversible, and transient. As a plastic feature, expression levels are modified through non-mutational processes upon changes in the environment. Therefore, efflux pumps are induced or up-regulated when a specific environmental cue, e.g. antibiotic concentration, is present. Several anti-TB drugs have been shown to induce the expression of efflux pump genes, and several transcription regulators of efflux pump genes have been described in Mycobacteria [61-63].
Mycobacterial efflux pumps are capable of extruding almost all anti-TB drugs including S, R, H, CFZ, FQs, E and even the recently introduced BDQ [64,65]. The number of efflux pumps in MTB is one of the largest among bacteria, considering the size of its genome. Drug efflux via Mycobacteria pumps is therefore highly relevant when considering the treatment of MDR-TB patients, and may constitute an earlier step in the evolutionary process that leads to MTB resistance to high concentrations of anti-TB drugs [66,67]. The efflux confers low levels of resistance, which can result in exposure to sub inhibitory concentrations of an antimicrobial, and consequently increase the likelihood of the development of other resistance mechanisms. Eventually, drug efflux modifications may participate together with mutations associated with antimicrobial resistance to determine anti-TB drug resistance levels [67]. The relevance of efflux pumps for MTB drug resistance has been reviewed elsewhere [59,68].
Mutations Associated with Drug Resistance in MTB
MTB has particular characteristics that influence several aspects of its development and survival, including the presentation of resistance to drugs used against the infection [69]. The very composition of the cell wall constitutes a physical barrier to the action of certain classes of drugs, but on the other hand, it prevents the acquisition of resistance by genetic transfer between individuals, via plasmids and transposons, for example [69-71].
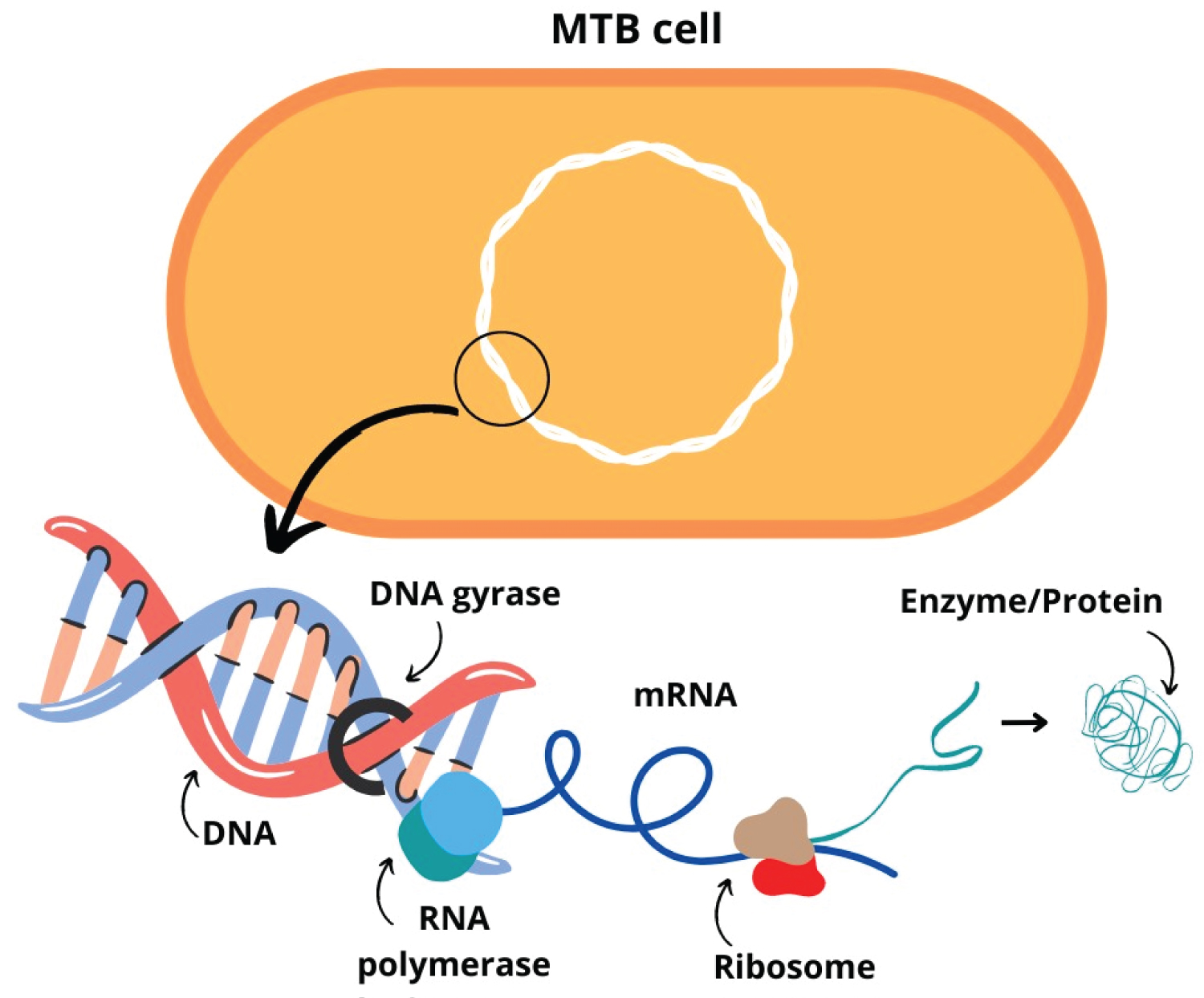
Thus, resistance acquisition by Mycobacterial depends on mutation events in genes involved in pathways relevant to the action of therapeutic drugs (Figure 3) [1]. These mutations are favored by the adverse environmental conditions resulting from the use of anti-TB drugs [70,72]. The mutation rate can reach 10 9 events per cell division round, under the selective pressure of therapy with anti-TB drugs. Both the nature of the drug and the strain under selective pressure influence the rate of mutagenesis [21].
DNA repair mechanisms are crucial for genome integrity. However, in the case of bacteria harboring mutations that confer resistance, the absence of repair mechanisms may be an additional advantage [21,73] associated alterations in Mycobacterial repair system genes, mut genes, with increased mutation rates in MTB strains of the Beijing family. The occurrence of mutations in DNA repair genes may be associated with suboptimal concentrations of anti-TB drugs [21].
Mutations can occur as single base polymorphisms (SNPs), deletions, or insertions, in one or multiple genes simultaneously [21]. Some mutations are more frequently represented causing a given resistance phenotype than others. For instance, the Ser315Thr mutation in the kat G gene found in H-resistant MTB [21,70] is considered a universal mutation associated with resistance to this drug [71,72]. The kat G gene encodes a catalase involved in the transformation of H into its active form [21,70,73]. Mutations in the inh A gene are also frequently present in H-resistant MTB. This gene encodes an NADH-reductase involved in the production of mycolic acid in the Mycobacterial wall [73]. Although these are the gene mutations most associated with H resistance, mutations in other genes, such as kas A, ahp C, and drf A, were also discovered [21,74-76].
Mutations in genes involved in the mechanism of action of the other first-line anti-TB drugs are also well studied. The rpo B gene encodes the Mycobacterial RNA polymerase β chain, the target of R [77]. Mutations in this gene are found in more than 90% of RR-TB strains [78]. Although these mutations can occur in different regions of the gene, they are primarily located in codons 507-533 [21]. For instance, the Ser531Leu mutation is present in the majority of resistant isolates from several countries including Brazil, Pakistan and China Reviewed in [77]. Mutations in pnc A are responsible for resistance to Z. This gene encodes a nicotinamidase (pyrazinamidase), which converts Z to pyrazinoic acid and contributes to the destabilization of MTB membrane transport. Pyrazinamide is a crucial anti-TB drug, as it acts on quiescent intracellular bacilli, contributing to shortening treatment time, in addition to acting on MTB resistant to R and H [1,79] The emb CAB operon encodes an arabinosyl transferase, which plays an important role in cell wall production. Mutations in emb B are associated with resistance to E, especially the one that occurs in codon 306 [21,23,80].
For second-line drugs, as well as for the new classes of drugs used in TB treatment, the occurrence of mutations that contribute to MTB resistance has also been reported. A variety of mutations have been mapped involving resistance to virtually all drugs available for TB treatment [21,23,81].
Epistasis and Compensatory Mutations
Epistasis can be defined, in a simplified way, as the modulation of phenotypic characteristics in a frame of gene interactions [82]. The concept is also applicable to the concurrence of multiple mutations that impact the survival capacity of organisms, as already demonstrated for MTB [72,83]. The result of this gene interaction varies according to the bacterial genetic background [83].
Mutations that confer resistance to antibiotics have consequences on the development of MTB. The so-called “fitness cost” interferes with Mycobacterial survival, growth, and virulence [62,72,76]. Additional mutations may compensate for fitness losses generated by resistance mutations, an example of the phenomenon of epistasis. These secondary mutations allowing for the restoration of Mycobacterial fitness act as compensatory mechanisms, and occur in genes that encode either the same proteins or proteins that participate in similar metabolic pathways, being associated with the maintenance of resistance to multiple drugs [21,69]. The simultaneous presence of mutations in different genes involved in resistance to a given drug can also contribute to the reduction of the fitness cost for MTB [69]. Epistatic relationships in MTB mutations also contribute to the maintenance of resistance after the withdrawal of drug pressure, as well as to the emergence of the multidrug resistance phenotype [69,72,83].
Post-Translational Changes and Mimicry of Pharmacological Targets
MTB drug resistance may also emerge from structural interference with the pharmacological targets of anti-TB drugs. The interaction between the drug and its target may be prevented by modifications in drug action sites, or by molecules that mimic these targets [69,71]. These epigenetic modifications can occur by different mechanisms, such as phosphorylation, glycosylation, methylation, acetylation, pupylation, biotinylation and phosphopantetheinylation [20,84,85].
MTB encodes an rRNA methyltransferase, capable of methylating the 23S portion of the rRNA, preventing the interaction between macrolide antibiotics and the ribosome [86]. The MTB-encoded TlyA methyltransferase alters the 50S ribosomal subunit, impairing the interaction of capreomycin with its target [87]. MTB DNA methylation is one of the mechanisms associated with para-aminosalicylic acid resistance [88].
The Mycobacterial fluoroquinolone resistance protein (MfpA) is an example of target mimicry. It has the ability to mimic the size, shape and surface of MTB DNA double helix and binds to DNA gyrase, preventing the action of FQs [89].
Conclusion
MTB presents many effective mechanisms to evade the action of anti-TB drugs. Such mechanisms can be intrinsic, such as the characteristic composition of its cell wall and the use of drug efflux pumps, or acquired, e.g. by mutations in genes that encode molecules involved in the action of anti-TB drugs, or in genes that compensate for fitness cost. The sites of drug action may be masked by structural modifications of the target molecules. MTB is also capable of producing molecules that mimic pharmacological targets, preventing direct action on such molecules. The occurrence of resistance in MTB is related to genetic aspects of the Mycobacterial, but also to environmental pressures, such as those induced by treatment, and may also be influenced by the genetic aspects of the host. Furthering the knowledge of MTB drug escape mechanisms and the surveillance of mutations in circulating strains is essential to the design of strategies that may allow for better detection of drug resistance and improved treatment outcomes.
References
- World Health Organization (2022) Who. Global Tuberculosis Report, 68 P.
- Kanabalan RD (2021) Human tuberculosis and Mycobacterium tuberculosis complex: A review on genetic diversity, pathogenesis and omics approaches in host biomarkers discovery. Microbiol Res 246: 126674.
- Daniel TM (2006) The history of tuberculosis. Respir Med 100: 1862-1870.
- Barberis I, Bragazzi NL, Galluzzo L, et al. (2017) The history of tuberculosis: From the first historical records to the isolation of Koch's bacillus. J Prev Med Hyg 58: E9-E12.
- Mohr KI (2016) History of antibiotics research. How to Overcome the Antibiotic Crisis. Curr Top Microbiol Immunol 398: 237-272.
- Keshavjee S, Farmer PE (2012) Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med 367: 931-936.
- Pontali E, Raviglione MC, Migliori GB, et al. (2019) Regimens to treat multidrug-resistant tuberculosis: Past, present and future perspectives. European Respiratory Review 28: 152190035.
- World Health Organization (1994) Who Tb, A Global Emergency.
- World Health Organization (2022) Who Consolidated Guidelines on Tuberculosis. Module 4: Treatment - Drug-Susceptible Tuberculosis Treatment.
- Monedero-Recuero I (2021) Situational analysis of 10 countries with a high burden of drug-resistant tuberculosis 2 years post-UNHLM declaration: Progress and setbacks in a changing landscape. Int J Infect Dis 108: 557-567.
- Allué-Guardia A, García JI, Torrelles JB, et al. (2021) Evolution of drug-resistant mycobacterium tuberculosis strains and their adaptation to the human lung environment. Front Microbiol 12: 612675.
- World Health Organization (2020) Guidance for the Surveillance of Drug Resistance in Tuberculosis, Sixth Edition. Geneva: World Health Organization.
- World Health Organization (2018) Who Technical Manual for Drug Susceptibility Testing Of Medicines Used in the Treatment of Tuberculosis.
- Acharya (2020) Advances in diagnosis of Tuberculosis: An update into molecular diagnosis of Mycobacterium tuberculosis. Mol Biol Rep 47: 4065-4075.
- Morel F (2020) Place de la biologie moléculaire dans le diagnostic de la tuberculose. Revue des Maladies Respiratoires 37: 412-416.
- Perez-Garcia F (2017) Diagnostic performance of Anyplex II MTB/MDR/XDR for detection of resistance to first and second line drugs in Mycobacterium tuberculosis. Microbiol Methods 139: 74-78.
- Meehan CJ (2019) Whole genome sequencing of Mycobacterium tuberculosis: Current standards and open issues. Nat Rev Microbiol 17: 533-545.
- World Health Organization (2022) Who. Global Tuberculosis Programme. 72.
- Crofton J, Mitchison DA (1948) Streptomycin resistance in pulmonary tuberculosis. Br Med 2: 1009-1015.
- Arora G (2021) Role of post-translational modifications in the acquisition of drug resistance in Mycobacterium tuberculosis. FEBS J 288: 3375-3393.
- Dookie N (2018) Evolution of drug resistance in Mycobacterium tuberculosis: A review on the molecular determinants of resistance and implications for personalized care. J Antimicrob Chemother 73: 1138-1151.
- Fonseca JD, Knight GM, Mchugh TD, et al. (2015) The complex evolution of antibiotic resistance in Mycobacterium tuberculosis. Int J Infect Dis 32: 94-100.
- Abraham (2020) Mechanism of Drug Resistance in Mycobacterium tuberculosis. Am J Biomed Sci Res 7: 378-383.
- Seung KJ, Keshavjee S, Rich ML, et al. (2015) Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb Perspect Med 5: a017863.
- World Health Organization (2022) Who Consolidated Guidelines on Tuberculosis. Module 4: Treatment - Drug-Resistant Tuberculosis Treatment, 2022 Update.
- Vanino E, Granozzi B, Akkerman OW, et al. (2023) Update of drug-resistant tuberculosis treatment guidelines: A turning point. International Journal of Infectious Diseases 130: S12-S15.
- Senghore M (2020) Evolution of Mycobacterium tuberculosis complex lineages and their role in an emerging threat of multidrug resistant tuberculosis in Bamako Mali. Sci Rep10: 327.
- Comas Al (2013) Out-of-Africa migration and Neolithic co expansion of Mycobacterium tuberculosis with modern humans. Nat Genet 45: 1176-1182.
- Brites D, Gagneux S (2017) The Nature and Evolution of Genomic Diversity in the Mycobacterium tuberculosis Complex. Adv Exp Med Biol 1-26.
- Negrete-Paz A, Vásquez-Marrufo M, Vázquez-Garcidueñas MS (2021) Whole-genome comparative analysis at the lineage/sublineage level discloses relationships between Mycobacterium tuberculosis genotype and clinical phenotype. Peer J 8: 12128.
- Coll F, Phelan J, Hill-Cawthorne GA, et al. (2018) Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat Genet 50: 307-316.
- Emane AKA (2021) Highly transmitted M. tuberculosis strains are more likely to evolve MDR/XDR and cause outbreaks, but what makes them highly transmitted? Tuberculosis 129: 102092.
- Brennan PJ, Nikaido H (1995) The envelope of mycobacteria. Annu Rev Biochem 64: 29-63.
- Elings W (2021) Two β-lactamase variants with reduced clavulanic acid inhibition display different millisecond dynamics. Antimicrob Agents Chemotherapy 65: 802628-20.
- Jarlier V, Nikaido H (1994) Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol Lett 123: 11-18.
- Nguyen L, Pieters J (2009) Mycobacterial subversion of chemotherapeutic reagents and host defense tactics: Challenges in tuberculosis drug development. Annu Rev Pharmacol Toxicol 49: 427-453.
- Sarathy JP, Dartois V, Lee EJD, et al. (2012) The role of transport mechanisms in Mycobacterium tuberculosis drug resistance and tolerance. Pharmaceuticals 5: 1210-1235.
- Ando H, Kitao T, Miyoshi-Akiyama T, et al. (2011) Downregulation of katG expression is associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol 79: 1615-1628.
- Cardoso (2007) Characterization of ndh gene of isoniazid resistant and susceptible Mycobacterium tuberculosis isolates from Brazil. Memórias do Instituto Oswaldo Cruz 102: 59-61.
- XU Y (2015) Mutations Found in embCAB, embR, and ubiA Genes of Ethambutol-Sensitive and -Resistant Mycobacterium tuberculosis Clinical Isolates from China. BioMed Research International 2015: 951706.
- Stephan J (2004) Multidrug resistance of a porin deletion mutant of Mycobacterium smegmatis. Antimicrob Agents Chemother 48: 4163-4170.
- Viveiros M (2012) Inhibitors of mycobacterial efflux pumps as potential boosters for anti-tubercular drugs. Expert Rev Anti Infect Ther 10: 983-998.
- Goossens SN, Sampson SL, Van Rie A, et al. (2020) Mechanisms of drug-induced tolerance in Mycobacterium tuberculosis. Clin Microbiol Rev 34: ee00141-20.
- Abraham (1941) Further observations on penicillin. The Lancet 177-189.
- Wang F, Cassidy C, Sacchettini JC, et al. (2006) Crystal structure and activity studies of the Mycobacterium tuberculosis β-lactamase reveal its critical role in resistance to β-lactam antibiotics. Antimicrob Agents Chemother 50: 2762-2771.
- Gonzalo X, Drobniewski F (2013) Is there a place for β-lactams in the treatment of multidrug-resistant/extensively drug-resistant tuberculosis? Synergy between meropenem and amoxicillin/clavulanate. J Antimicrob Chemother 68: 366-369.
- Hugonnet J, Blanchard JS (2007) Irreversible inhibition of the Mycobacterium tuberculosis β-lactamase by clavulanate. Biochemistry 46: 11998-12004.
- Cordillot M (2013) In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by L, D-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob Agents Chemother 12: 5940-5945.
- Dubée V (2012) Inactivation of Mycobacterium tuberculosis L, D-transpeptidase LdtMt1 by carbapenems and cephalosporin’s. Antimicrob Agents Chemother 56: 4189-4195.
- Kumar P, Arora K, Lloyd JR, et al. (2012) Meropenem inhibits D,D-carboxypeptidase activity in Mycobacterium tuberculosis. Mol Microbiol 86: 367-381.
- Elings W (2021) Two β-lactamase variants with reduced clavulanic acid inhibition display different millisecond dynamics. Antimicrob Agents Chemother 65: 802628-20.
- SOROKA D (2015) Hydrolysis of clavulanate by Mycobacterium tuberculosis β-lactamase BlaC harboring a canonical SDN motif. Antimicrob Agents Chemother 59: 5714-5720.
- Van Alen I (2021) The G132S mutation enhances the resistance of Mycobacterium tuberculosis β-lactamase against sulbactam. Biochemistry 60: 2236-2245.
- Warrier T (2015) Identification of Novel Anti-mycobacterial Compounds by Screening a Pharmaceutical Small-Molecule Library against Nonreplicating Mycobacterium tuberculosis. ACS Infect Dis 1: 580-585.
- Warrier T (2016) N-methylation of a bactericidal compound as a resistance mechanism in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America 31: 4523-4530.
- Houghton JL (2013) Unexpected N-acetylation of capreomycin by mycobacterial Eis enzymes. J Antimicrob Chemother 68: 800-805.
- Pang AH (2022) Discovery of substituted benzyloxy-benzylamine inhibitors of acetyltransferase Eis and their anti-mycobacterial activity. Eur J Med Chem 242: 114698.
- Zaunbrecher MA, Sikes RD Jr, Metchock B, et al. (2009) Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences 106: 20004-20009.
- Kambli P (2016) Correlating rrs and eis promoter mutations in clinical isolates of Mycobacterium tuberculosis with phenotypic susceptibility levels to the second-line injectables. Int J Mycobacteriol 5: 1-6.
- Kanji A, Hasan R, Hasan Z, et al. (2019) Efflux pump as alternate mechanism for drug resistance in Mycobacterium tuberculosis. Indian J Tuberc 66: 20-25.
- Dulberger CL, Rubin EJ, Boutte CC, et al. (2020) The mycobacterial cell envelope-a moving target. Nat Rev Microbiol 18: 47-59.
- Bolla JR (2012) Structural and functional analysis of the transcriptional regulator Rv3066 of Mycobacterium tuberculosis. Nucleic Acids Res 40: 9340-9355.
- Da Silva (2011) Efflux as a mechanism for drug resistance in Mycobacterium tuberculosis. FEMS Microbiol Immunol 1-9.
- Vadija R (2018) Identification of small molecular inhibitors for efflux protein Rv2688c of Mycobacterium tuberculosis. Biotechnol Appl Biochem 65: 608-621.
- Andries (2014) Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9: e102135.
- Almeida (2016) Mutations in pepQ confer low-level resistance to bedaquiline and clofazimine in Mycobacterium tuberculosis. Antimicrob Agents Chemother 60: 4590-4599.
- Kardan-Yamchi J (2019) Expression analysis of 10 efflux pump genes in multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis clinical isolates. J Glob Antimicrob Resist 17: 201-208.
- Machado D (2012) Contribution of efflux to the emergence of isoniazid and multidrug resistance in Mycobacterium tuberculosis. PLoS One 7: e34538.
- Te Brake LHM (2018) The Role of Efflux Pumps in Tuberculosis Treatment and Their Promise as a Target in Drug Development: Unraveling the Black Box. Annu Rev Pharmacol Toxicol 6: 271-291.
- Gygli SM (2017) Antimicrobial resistance in Mycobacterium tuberculosis: Mechanistic and evolutionary perspectives. FEMS Microbiol Rev 41: 354-373.
- Swain SS (2020) Molecular mechanisms of underlying genetic factors and associated mutations for drug resistance in Mycobacterium tuberculosis. Emerg Microbes Infect 9: 1651-1663.
- Al-Saeedi M, Al-Hajoj S (2017) Diversity and evolution of drug resistance mechanisms in Mycobacterium tuberculosis. Infect Drug Resist 10: 333-342.
- Singh R (2019) Recent updates on drug resistance in Mycobacterium tuberculosis. J Appl Microbiol 128: 1547-1567.
- Rad ME (2003) Mutations in Putative Mutator Genes of Mycobacterium tuberculosis Strains of the W-Beijing Family. Emerg Infect Dis 9: 838-845.
- Marahatta SB (2011) katG (SER 315 THR) Gene Mutation in Isoniazid Resistant Mycobacterium tuberculosis. Kathmandu Univ Med J 32: 19-23.
- Gazzi (2017) TP Synthesis, Inhibition of Mycobacterium tuberculosis Enoyl-acyl Carrier Protein Reductase and Antimycobacterial Activity of Novel Pentacyanoferrate(II)-isonicotinoylhydrazones. J Braz Chem Soc 10: 2028-2037.
- Da Silva PEA, Palomino JC (2011) Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: Classical and new drugs. J Antimicrob Chemother 66: 1417-1430.
- Zaw MT, Emran NA, Lin Z, et al. (2018) Mutations inside rifampicin-resistance determining region of rpoB gene associated with rifampicin-resistance in Mycobacterium tuberculosis. J Infect Public Health 11: 605-610.
- Damtie D, Woldeyohannes D, Mathewos B, et al. (2014) Review on Molecular mechanism of first line antibiotic resistance in Mycobacterium tuberculosis. Mycobact Dis 4: 6.
- Njire M (2015) Pyrazinamide resistance in Mycobacterium tuberculosis: Review and update. Adv Med Sci 125: 1-9.
- Plinke C (2009) Tuberculosis ethambutol resistance: Concordance between phenotypic and genotypic test results. Tuberculosis 89: 448-452.
- The Cryptic Consortium (2022) A data compendium associating the genomes of 12,289 Mycobacterium tuberculosis isolates with quantitative resistance phenotypes to 13 antibiotics. Plos Biology 9: 3001721.
- Domingo J, Baeza-Centurion P, Lehner B, (2019) The Causes and Consequences of Genetic Interactions (Epistasis). Annu Rev Genomics Hum Genet 20: 433-460.
- Borrell S, Gagneux S (2011) Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin Microbiol Infect 17: 815-820.
- Chen L, Li H, Chen T, et al. (2018) Genome-wide DNA methylation and transcriptome changes in Mycobacterium tuberculosis with rifampicin and isoniazid resistance. Int J Clin Exp Pathol 11: 3036-3045.
- Richard-Greenblatt M, Av-Gay Y (2017) Epigenetic Phosphorylation Control of Mycobacterium tuberculosis Infection and Persistence. Microbiol Spectr 5.
- Nguyen L (2016) Antibiotic resistance mechanisms in M. tuberculosis: An update. Arch Toxicol 90: 1585-1604.
- Laughlin Z (2022) 50s subunit recognition and modification by the Mycobacterium tuberculosis ribosomal RNA methyltransferase TlyA. PNAS Biochemistry 119: 14.
- LI XM (2019) Synthesis and structure-bactericidal activity relationships of non-ketolides: 9-Oxime clarithromycin 11,12-cyclic carbonate featured with three-to eight-atom-length spacers at 3-OH. Eur J Med Chem 171: 235-254.
- Gautam P, Shivangi, Meena LS, et al. (2020) Revelation of point mutations effect in Mycobacterium tuberculosis MfpA protein that involved in mycobacterial DNA supercoiling and fluoroquinolone resistance. Biotechnol Appl Biochem 68: 1357-1371.
Corresponding Author
Clarissa Cunha Santana, Departamento de Ciências Biológicas, Universidade Estadual do Sudoeste da Bahia, Rua José Moreira Sobrinho, 45206-190, Jequiezinho, Jequie, Bahia, Brazil.
Copyright
© 2023 Santana CC, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.