Vogt-Koyanagi-Harada Syndrome: A Case of Bilateral Steroid-Responsive Loss of Vision
Abstract
We describe a 35-year-old Caucasian woman with a bilateral loss of vision progressed over 10 days, showing optic disk oedema at examination. Serological screening and Cerebrospinal Fluid (CSF) analysis excluded any infective, neoplastic, granulomatous or rheumatologic disorder. Axial brain magnetic resonance imaging showed a bilateral thickening of the choroid with retinal detachments, and few small cerebral periventricular white matter lesions. An ophthalmological review confirmed serous retinal detachments, and performed a retinal fluorescein angiography. These findings suggested an uncommon uveo meningitic disease, the Vogt-Koyanagi-Harada (VKH) syndrome. This rare autoimmune condition classically includes bilateral uveitic manifestations associated with neurological, auditory and skin involvement. When clinical presentation is incomplete, VKH syndrome diagnosis is supported by ocular and brain imaging to start an early treatment, as well as CSF analysis. High-dose steroids or immunosuppressant are the gold-standard treatments: Our patient improved quickly after intravenous methylprednisolone but worsened during tapering so we added Azathioprine, also as steroid-sparing agent, obtaining a complete and permanent recovery.
Keywords
Vogt-Koyanagi-Harada syndrome, Bilateral loss of vision, Steroid-responsive autoimmune loss of vision
Background
Vogt-Koyanagi-Harada (VKH) syndrome is a rare autoimmune disorder related to a T-lymphocyte-mediated response against melanocyte tyrosinase-related proteins. The onset phenotype is generally an uveo meningitic syndrome, with associated hearing loss and skin involvement [1]. Diagnostic criteria include: Bilateral choroiditis with bullous serous retinal detachments or focal areas of subretinal fluid, neurological/auditory symptoms (meningismus, tinnitus), and integumentary features (alopecia, poliosis, vitiligo) [2]. If the clinical presentation is incomplete, ocular and brain imaging could support an early diagnosis. Prompt treatment with steroids and other immunosuppressive drugs is needed to prevent disease progression and recurrences [3].
Case Presentation
Our patient is a 35-year-old Caucasian woman from Albany who presented a bilateral loss of vision that progressed over 10 days, and a mild tension-type headache. Previous medical history was unremarkable, and she denied any history of ocular trauma. Family history was mute.
Neurological examination was normal except for bilateral asymmetrical optic disk oedema and mild auditory sensorial deficits. There was Neither meningismus nor signs of brainstem involvement.
After an unspecific brain computerized tomography, we performed a spinal tap to exclude intracranial hypertension, and the opening pressure was normal. Cerebrospinal Fluid (CSF) analysis showed mild pleocytosis (10 cells/mm3, all lymphocytes), normal proteins, and no oligoclonal bands. CSF cytopathology and infective work-up for herpetic virus were negative. Blood tests were unremarkable, except for the erythrocyte sedimentation rate elevated to 20 mm/hr. We tested for: Tuberculosis (quantiferon was negative), Lyme disease, syphilis, human immunodeficiency virus, lymphoproliferative disorders (normal blood cytofluorometry), Sarcoidosis (serum angiotensin-converting enzyme), Lupus erythematous (antinuclear antibody), and Wegener's granulomatosis (antineutrophil cytoplasmic antibodies). Chest x-ray was normal (excluding signs of sarcoidosis too), and the patient denied any history of mucosal ulceration (suggestive for Behcet's disease).
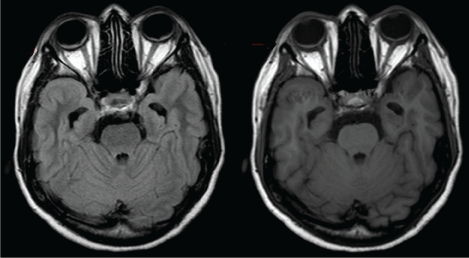
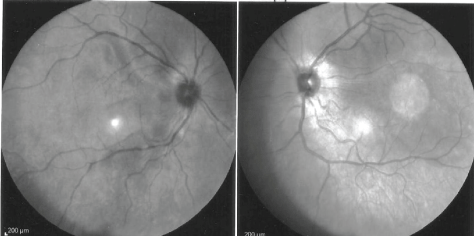
Since we were first suspecting for a neurological disorder, brain Magnetic Resonance (MR) imaging was performed and the axial T2 and T1-sequences showed a bilateral thickening of the choroid, and few small periventricular white matter lesions with no contrast-enhancement (Figure 1). We then asked an ophthalmological review confirming bilateral serous retinal detachments (not clearly seen at first bed-examination funduscopy, and slit lamp eye exam was not performed at admission), and the Optical Coherence Tomography (OCT) evidenced areas of subretinal fluid. B-scan ultrasonography was not performed. These findings suggested a granulomatous panuveitis, and, subsequently, our patient underwent a retinal fluorescein angiography showing bilateral serous retinal detachments too (Figure 2). The clinical history, CSF pleocytosis, the brain MR imaging, and the ocular findings (including OCT and retinal angiography) were suggestive for VKH syndrome. We started intravenous methyprednisolone 1 g daily for 5 days, and our patient completely recovered both the vision and the hearing loss. We tapered with oral prednisolone, but, at 20 mg daily two months later, she complained again of blurred vision. We then rose up steroids to 50 mg, and started Azathioprine up to 150 mg daily: The patient recovered completely after one month. Subsequently we progressively stopped steroids with no further recurrences. Azathioprine has been effective and well-tolerated. Our patient has been asymptomatic for 3 years after the VKH onset, and ophthalmological periodical reviews have been negative too.
Discussion
We presented a young Albanian woman with bilateral vision loss associated with retinal serous detachments, sensorial hearing deficit and mild headache. She had no skin involvement (no poliosis, vitiligo or alopecia), and the examination excluded meningismus and brainstem dysfunction. This clinical picture fits with incomplete VKH syndrome, according to diagnostic criteria revised in 2007. A "complete" form of VKH is less frequent in the early phases of the disease, and skin is commonly spared at first symptoms, as happened in our case. On the other hand, VKH is rare in Caucasian ethnicity being more frequent in Asians [2].
Incomplete VKH could mimic neurological disorders, in fact our first thoughts have been intracranial benign hypertension (excluded with the spinal tap opening pressure), and infections (excluded with the CSF analysis). Obviously, eye features are fundamental to suspect VKH, but rachicentesis is useful to support this hypothesis since headache alone does not define "the typical neurological involvement" or meningismus, whereas a mild CSF pleocytosis does. It is also this latter CSF finding with the presence of multifocal serous retinal detachments that suggested us to focus on uveo meningeal syndromes. Subsequent differential diagnosis was initially ruled out with blood tests for infective, neoplastic, granulomatous or rheumatologic disorders.
Another clue for us to diagnose VKH has been, unexpectedly, MR imaging with signs of choroiditis and retinal detachments. Unspecific brain abnormalities have been reported in VKH too: Pachymeningeal enhancement, few scattered cerebral periventricular white matter lesions with no enhancement, or within the brainstem and peduncles [4,5].
Finally, an expert ophthalmological review is necessary to confirm ocular findings of VKH performing also OCT and retinal fluorescein angiography [3].
Immediately after the diagnostic work up, high-dose intravenous steroids or immunosuppressant must be started to obtain a complete recovery even in an incomplete early phase of VKH [6]. Some patients could relapse when tapering with oral treatment steroids, or have some side-effects related to long-term steroids, thus suggesting the use of immunosuppressive therapies since the early course: Cyclosporine, mycophenolate, and azathioprine are similarly effective [7,8]. Our patient had a complete recovery of the visual acuity with high-dose steroids, she relapsed when it was tapered, and remained disease-free with azathioprine.
Acknowledgement
Many thanks to Dr. Elisa Buschini, Ophthalmologists and friend.
Conflict of Interest
All the authors have no disclosure.
References
- Sakata VM, da Silva FT, Hirata CE, et al. (2014) Diagnosis and classification of Vogt-Koyanagi-Harada disease. Autoimmun Rev 13: 550-555.
- Rao N, Sukavatcharin S, Tsai J (2007) Vogt-Koyanagi-Harada disease diagnostic criteria. Int Ophthalmol 27: 195-199.
- Abu El-Asrar AM, Al Tamimi M, Hemachandran S, et al. (2013) Prognostic factors for clinical outcomes in patients with Vogt-Koyanagi-Harada disease treated with high-dose corticosteroids. Acta Ophthalmol 91: e486-e493.
- Baltmr A, Lightman S, Tomkins-Netzer O (2016) Vogt-Koyanagi-Harada syndrome - current perspectives. Clin Ophthalmol 10: 2345-2361.
- Keles S, Ogul H, Pinar LC, et al. (2013) Teaching neuroimages: cerebral white matter involvement in a patient with Vogt-Koyanagi-Harada syndrome. Neurology 81: e85-e86.
- Lohman BD, Gustafson CA, McKinney AM, et al. (2011) MR imaging of Vogt-Koyanagi-Harada syndrome with leptomeningeal enhancement. AJNR Am J Neuroradiol 32: E169-E171.
- Greco A, Fusconi M, Gallo A, et al. (2013) Vogt-Koyanagi-Harada syndrome. Autoimmun Rev 12: 1033-1038.
- Arcinue CA, Radwan A, Lebanan MO, et al. (2013) Comparison of two different combination immunosuppressive therapies in the treatment of Vogt-Koyonagi-Harada syndrome. Ocul Immunol Inflamm 21: 47-52.
Corresponding Author
Domizia Vecchio, MD, Neurological Department, AOU Maggiore della Carità and A. Avogadro University of Piemonte Orientale, Corso Mazzini 18, 28100 Novara, Italy, Tel: 0321 373 3964.
Copyright
© 2017 Vecchio D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.