Sinonasal Rhabdomyosarcoma in a Child: Case Report and Review of the Literature
Abstract
Rhabdomyosarcoma is a malignant soft-tissue neoplasm of skeletal muscle origin, which predominantly affects the young population. Embryonal and alveolar are the most common histological subtypes of rhabdomyosarcoma. We present a case of a 3-year-old child with alveolar rhabdomyosarcoma arising from the left nasal cavity. CT revealed a mass lesion with a marked contrast enhancement in the left nasal cavity and immunohistochemical studie was conducted to further classify this lesion. Neoplastic cells were positive for myogenin. We managed the patient with surgical resection followed by radiotherapy.
Keywords
Rhabdomyosarcoma, Immunohistochemical, Nasal cavity
Introduction
Rhabdomyosarcoma (RMS) is the sarcoma of the soft tissue originating from skeletal muscle. The most common in children, adolescent and adults young. Its preferred locations are the head and neck in 44% of cases [1].
Diagnosing RMS is difficult with histology alone. Immunostaining is one of the most important methods that can lead to a specific diagnosis [2]. Treatment of RMS is based on risk stratification at the time of diagnosis. Surgical removal of the tumor and chemotherapy or combination of both is the treatment of choice. The current report illustrates the clinical and pathological features of alveolar RMS originating in the nasal cavity in a child.
Case Report
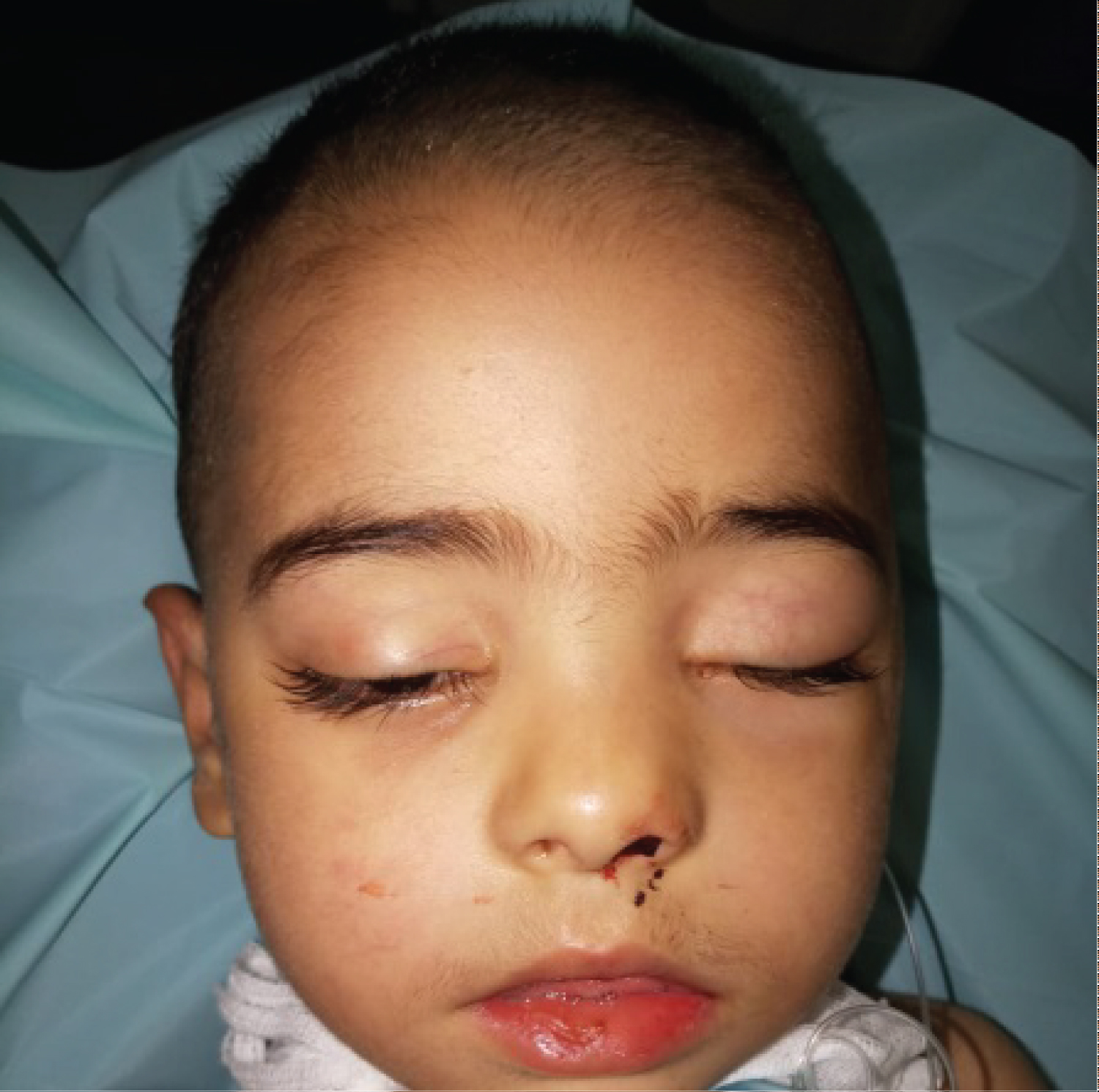
A 3-year-old moroccan boy with no relevant medical or family history, was admitted to our ENT department for the evaluation and treatment of chronic nasal obstruction and epistaxis evolving for 6 months. On referral, head and neck evaluation revealed a friable, purplish-colored tumor with an irregular surface, bleeding on contact, herniating into the left anterior vestibule, completely obstructing the left nasal cavity and descending under the soft palate to para-pharyngeal area. Right nasal cavity was free (Figure 1).
The rest of his head and neck examination, including cranial nerve evaluation, was normal.
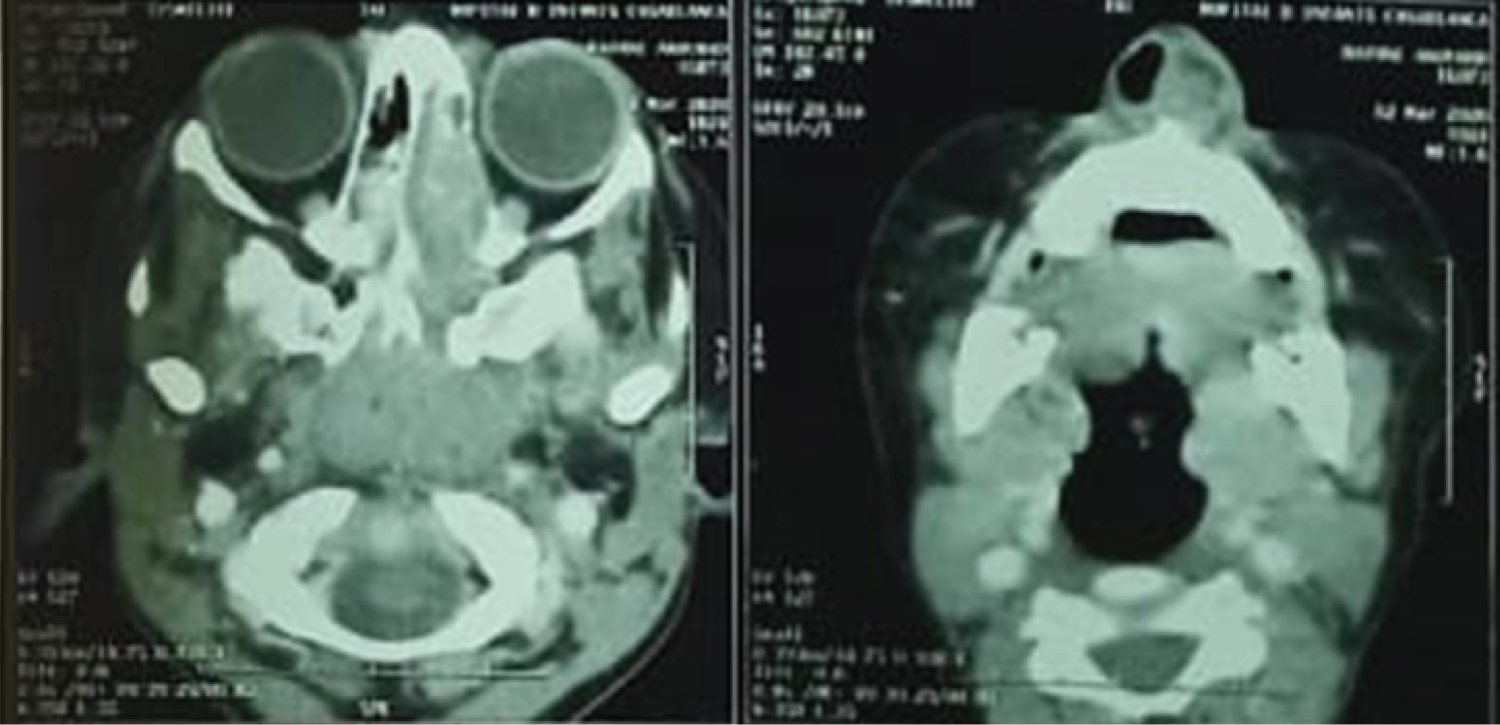
A sinus computed tomography (CT) scan was obtained showing a large mass filling the left nasal cavity, hypodense with heterogeneous enhaucement, measuring 75mmY*45mm * 35mm, extending posteriorly along the lateral nasal wall into the nasopharynx and the medial wall of the orbite without intra orbital invasion or endocranial extension. (Figure 2)
Based on these findings, the patient was taken to the operating room for biopsy. Histologic examination revealed large round cells with hyperchromatic nuclei and eosinophilic cytoplasm, immunohistochemical analysis was performed to further classify this lesion. The neoplastic cells were strongly positive for myogenin
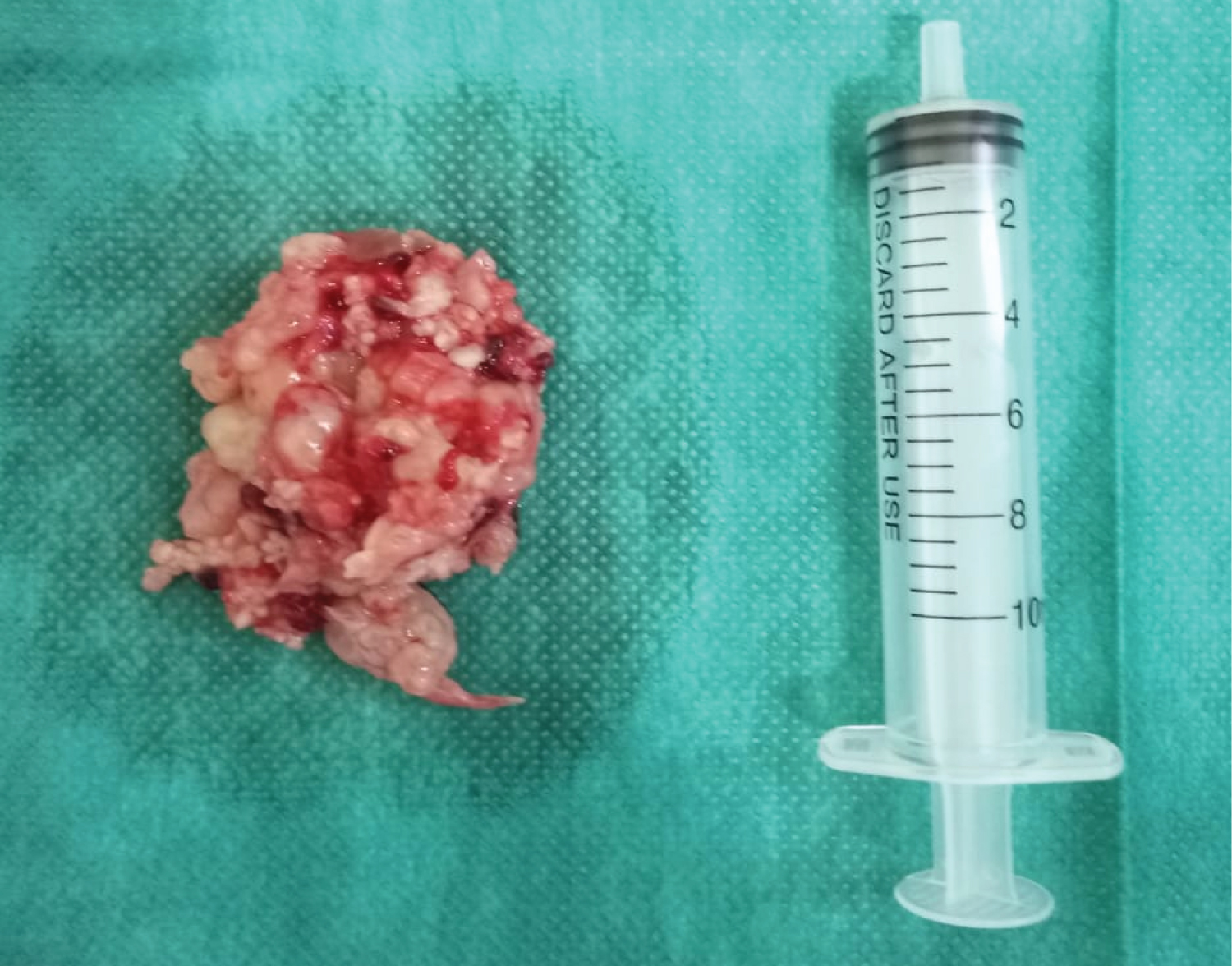
Under general anaesthesia, endoscopic sinus surgery was performed. The mass was removed en bloc (Figure 3)
The treatment was followed by radiotherapy.
Discussion
Tumours in the nasal cavity and paranasal sinus are rare, affecting less than 1 in 100 000 people per year [3], malignant are more common than their benign counterparts, they represente only 3% of all head and neck cancers [4].
Rhabdomyosarcoma (RMS) is a primitive malignant mesenchymal tumour that originates from immature cells that are destined to differentiate into striated skeletal muscle [5], accounting for 3-5% of all malignancies in childhood. Approximately 40% of all RMS occur in the head and neck region some 20% of which involve the sinonasal tract and nasopharynx [6,7].
Rhabdomyosarcoma falls into three main groups; embryonal, alveolar and pleomorphic, the first two groups occur mainly in children, while pure pleomorphic lesions occural most exclusively in adults [8].
The average age at diagnosis is 5-6 years, and the majority of patients are less than 10 years old; 4 without a sex predilection [6]. The presenting signs and symptoms of RMS are variable, depending on the site of initial presentation, the extent of the tumour and the presence or absence of distant metastases and lymph-node involvement. Patients generally present with nasal obstruction, epistaxis, facial swelling, exophthalmos, decreases in vision, proptosis and diplopia and sinusitis, result in blockage of sinus ostia [9].
Imaging appearence of the RMS is non-specific. The tumour may be illimited defined with infiltrative margins or well circumscribed by a pseudocapsule or compressed tissue. CT is helpful to evaluate bone erosion. MRI imaging is gives a better definition of the mass and its invasion of adjacent structures [10].
The histological appearance of alveolar RMS is characterizing by fibrous septa separating loosely cohesive rhabdomyoblasts into alveolar spaces; multinucleated giant, round tumor cells may be present with a typical alveolar appearance. The deep acidophilia of the cytoplasm is also important diagnostic features [11].
Histological presentation can make diagnosis difficult hence the benefit of complementing it with immunohistochemistry and additional genetic tests, immunohistochemical features in RMS is caractirezed by expression of desmin, myogenin, MYOD1, muscle-specific actin, smooth muscle actin, myoglobin, fast myosin, MITF, and CD56 the Myogenin is clearly the most sensitive and specific marker of rhabdomyosarcoma, it is expressed in a particularly strong and diffuse fashion in the alveolar type. In the genitic test this tumor is characterized by the recurrent translocations which involve the FOXO1A gene on chromosome 13 with either PAX3 on chromosome 2 or PAX7 on chromosome 1 [12].
The treatment of RMS is based on surgery, radiotherapy, chemotherapy according to the risk stratification at the time of diagnosis. Surgical excision should be complete with normal tissue margins of at least 0.5 cm surrounding the tumor. In cases in which complete excision is not possible and residual disease is left, the surgical resection should be followed by an earlier radiotherapy [13].
Radiotherapie along with surgical resection is an essential part of local control which an earlier start at radiotherapie confers better local control. Proton beam RT represents a safe and effective radiation modality for pediatric RMS patients with improved 5-year local control (81%), EFS (69%), and OS (78%) [14].
The use of multidrug chemotherapy and external beam radiation followed by surgery has resulted in a dramatic increase in survival for those children with rhabdomyosarcoma. The standard chemotherapy regimen includes vincristine, actinomycin-D, and cyclophosphamide (VAC) [15]. Adjuvant or neoadjuvant chemotherapy may be of interest in locally advanced and/or inoperable tumours.
A better prognosis is seen in children, the 5-year survival rate for patients with RMS has improved substantially to an over all survival (OS) of 70%- 75% in the recent Intergroup Rhabdomyosarcoma Study (IRS) and International Society of Pediatric Oncology (SIOP) study [16,17].
Conclusion
Any child presenting with suspicious symptoms requires a detailed imaging evaluation and, when indicated, a biopsy for histopathologic confirmation. Diagnosing rhabdomyosarcoma before it has spread extensively not only increases survival, but also allows less toxic treatments to be curative.
References
- Montone KT, Barr FG, Zhang PJ, et al. (2009) Embryonal and alveolar rhabdomyosarcoma of parameningeal sites in adults: a report of 13 cases. Int J Surg Pathol 17: 22-30.
- Wick MR, Swanson PE, Scheithauer BW, et al. (1987) Malignant peripheral nerve sheath tumor: An immunohistochemical study of 62 cases. Am J Clin Pathol 87: 425-433.
- Lund VJ, Howard D, Wei W (2014) Tumours of the nose, sinuses and nasopharynx. Thieme 1-595.
- Haerle SK, Gullane PJ, Witterick IJ, et al. (2013) Sinonasal carcinomas: Epidemiology, pathology, and management. Neurosurg Clin N Am 24 : 39-49.
- Cavazzana AO, Schmidt D, Ninfo V, et al. (1992) Spindle cell rhabdomyosarcoma: A prognostically favourable variant of rhabdomyosarcoma. Am J Surg Pathol 16: 229-235.
- Freling NJ, Merks JH, Saeed P, et al. (2010) Imaging findings in craniofacial childhood rhabdomyosarcoma. Pediatr Radiol 40 : 1723-1738.
- Justin A Bishop, Lester DR (2019) Thompson malignant neoplasms of the nasal cavity, paranasal sinuses, and nasopharynx. Head and Neck Pathology (Third Edition).
- Christopher DM Fletcher (2014) Recently characterized soft tissue tumors that bring biologic insight. Mod Pathol 27: 98-112.
- Goosens V, Van den Berghe I, CDe Clercq, et al. (2008) Radiation-induced mandibular adult spindle cell rhabdomyosarcoma. Int J Oral Maxillofac Surg 37: 395-397.
- Nascimento AF, Fletcher CD (2005) Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol 29: 1106-1113.
- John Goldblum, Laura Lamps, Jesse McKenney, et al. (2017) Soft Tissues. John R. Goldblum Rosai and Ackerman's Surgical Pathology, 41: 1810-1914.
- Leona A Doyle, Jason L Hornick (2019) Immunohistology of neoplasms of soft tissue and bone bone. Diagnostic Immunohistochemistry, Chapter 4, 82-136.e14.
- Blakely ML, Lobe T, Anderson J, et al. (1999) Does debulking improve survival rate in advanced-stage retroperitoneal embryonal rhabdomyosarcoma? J Pediatr Surg 34: 736-742.
- Ladra MM, Szymonifka J, Mahajan A, et al. (2014) Preliminary results of a phase II trial of proton radiotherapy for pediatric rhabdomyosarcoma. J Clin Oncol 32: 3762-3770.
- Oberlin O, Rey A, Anderson J, et al. (2001) Treatment of orbital rhabdomyosarcoma: Survival and late effects of treatment e results of an international workshop. J Clin Oncol 19: 197-e204.
- Meza JL, Anderson J, Pappo AS, et al. (2006) Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The Children’s Oncology Group. J Clin Oncol 24: 3844-3851.
- Raney RB, Anderson JR, Barr FG, et al. (2001) Rhabdomyosarcoma and undifferentiated sarcoma in the first two decades oflife: A selective review ofintergroup rhabdomyosarcoma study group experience and rationale for Intergroup Rhabdomyosarcoma Study V. J Pediatr Hematol Oncol 23: 215-220.
Corresponding Author
M Laachoubi, Department of Otolaryngology and Head and Neck Surgery, Hospital 20 August 1953, Morocco
Copyright
© 2022 Laachoubi M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.