Juvenile Aggressive Ossifying Fibroma of Paranasal Sinuses: An Unusual Presentation in an 18-Year-Old Female
Abstract
Juvenile aggressive ossifying fibroma (JAOF) is a rare variant of ossifying fibroma. Conventional ossifying fibromas are slow growing neoplasms thought to originate from osseous mesenchyme, commonly arising in the mandible. Conversely, JAOF occurs more frequently in the paranasal bones with rapid growth leading to local destruction, facial asymmetry, nasal obstruction, and proptosis. JAOF has two microscopic variants, psammomatoid and trabecular. Most JAOFs occur in patients less than 12-years-old with slight male predominance. We present an 18-year-old female with facial asymmetry for two years duration and recent onset of severe headaches. Examination revealed a bulging left maxillary bone and left upward proptosis. MRI showed an 81.2 × 44 × 39 mm, lobulated, well-defined, expansile osseous lesion with calcified matrix and soft tissue component involving the left maxillary sinus with extension into the left nasal cavity, frontal and ethmoid sinuses, and left orbit. An endoscopic biopsy procedure was performed to reveal sinonasal mucosa infiltrated by a cellular, fibro-osseous neoplasm consisting primarily of numerous, closely packed, round, uniform basophilic psammoma bodies with eosinophilic rings, interspersed by cellular, benign fibroblastic proliferations. Cystic changes and foci of necrosis were also noted. Complete excision of the JAOF is critical as recurrence and corresponding complications may develop in up to 50% of patients.
Keywords
Ossifying Fibroma, Spindle Cell, Calcification, Osteoblasts, Case Report
Introduction
Conventional ossifying fibromas (OF) are benign, slow growing neoplasms, typically found in the gnathic bones, and thought to originate from the osseous mesenchyme or the periodontal ligament [1,2]. These tumors typically present in females in their third and fourth decade of life, with a propensity for the teeth-bearing areas of the mandible or maxilla [3]. Conversely, juvenile aggressive ossifying fibromas (JAOF) typically occur in the paranasal sinuses and craniofacial bones with rapid growth leading to local destruction, facial deformity, and associated complications. There are two histologic variants of JAOF: Juvenile trabecular ossifying fibroma (JTOF) and juvenile psammomatoid ossifying fibroma (JPOF). Both entities are rare in comparison to conventional ossifying fibroma and are indistinguishable on imaging. Therefore, clinical, radiologic, and pathologic correlations are required to reach the correct diagnosis. Herein, we present a case of juvenile psammomatoid ossifying fibroma of the left craniofacial bones with extensive growth and expansion into the frontal and ethmoid sinuses, nasal cavity, orbit, and skull base.
Case Report
An 18-year-old African American female presented with facial asymmetry and prominent left facial swelling for two years duration. She recently experienced two severe headaches with little to no improvement with medications as well as loss in her sense of smell and blurred vision. Her past medical history is significant for late onset menarche and migraines. On examination, facial asymmetry with bulging left maxillary bone and left upward proptosis was noted. Extraocular muscle movement and cranial nerves II-XII were grossly intact and symmetrical. Rhinoscopy showed a markedly deviated nasal septum to the right without turbinate hypertrophy. Oropharyngeal examination showed good dentition, no loose teeth, and no palato-pharyngeal mass or bulge with fully mobile and symmetrical tongue and palate.
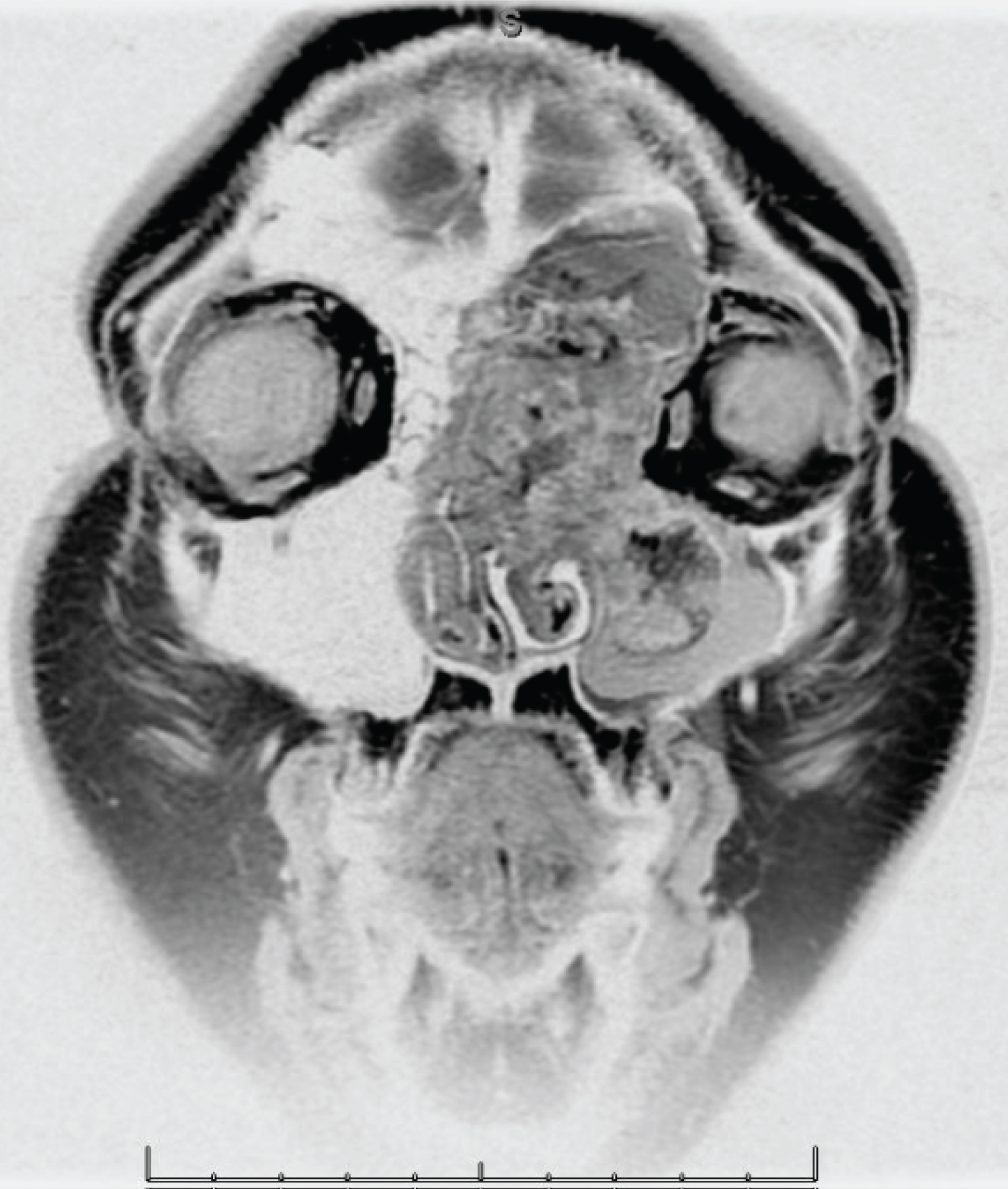
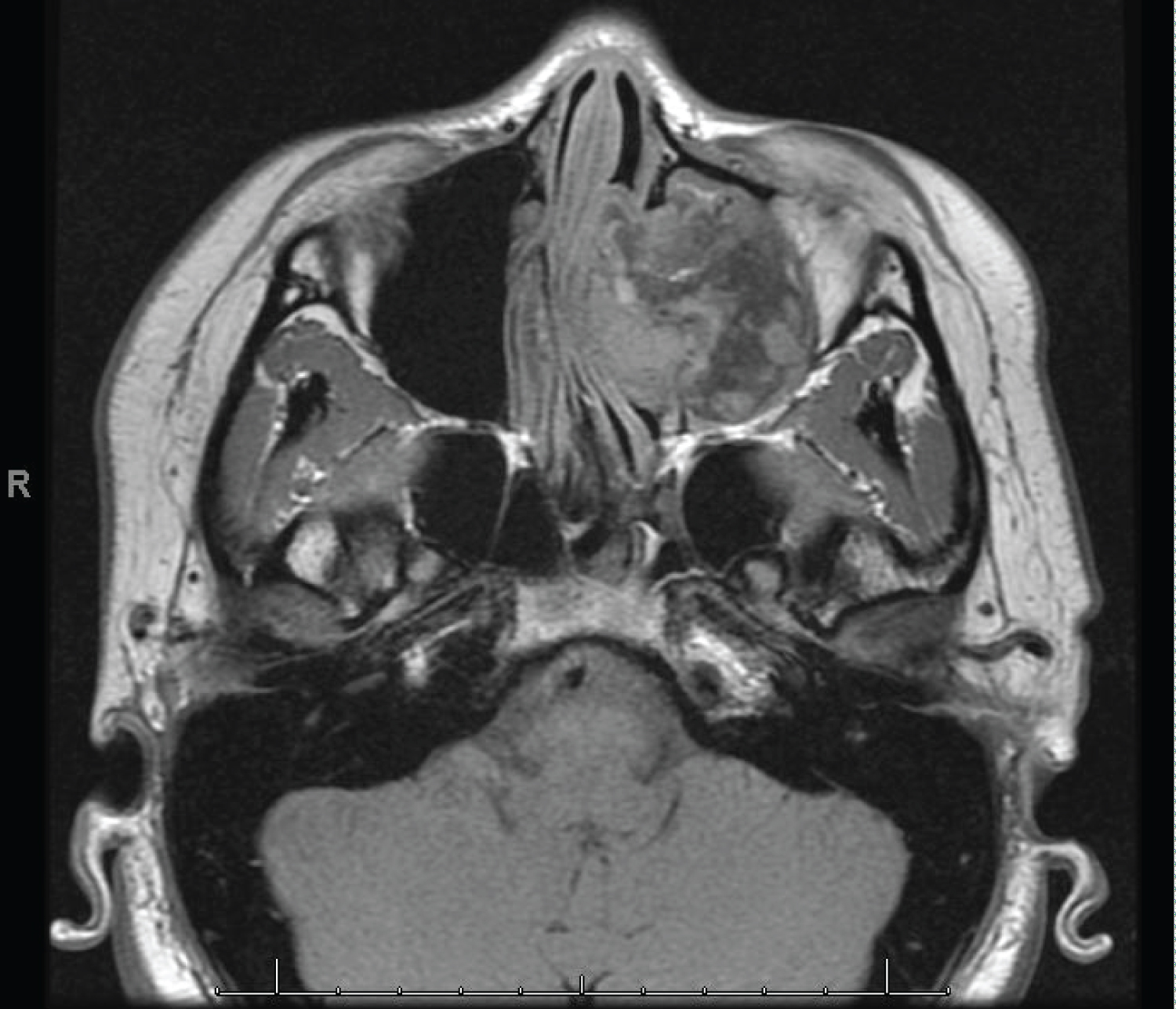
Initial CT showed an extradural mass involving the left nasal cavity and paranasal sinuses with mass effect on the left orbit and skull base without local destruction. An MRI was subsequently performed to reveal an 81.2 × 44 × 39 mm, lobulated, well-defined, heterogeneously enhanced, expansile osseous lesion with calcified matrix and soft tissue component of the left maxillary sinus with extension into the left nasal cavity, frontal and ethmoid sinuses, and medial wall of the left orbit. Deformity of the nasal septum, vomer, and medial and inferior walls of the orbit were noted with partial effacement of the left sphenopalatine fossa. Additionally, associated deformity at the left frontal rectus gyrus, left orbitofrontal gyrus, and left olfactory bulb were observed on imaging. Osteolysis at the left cribriform plate was not appreciated (Figure 1 and Figure 2).
Due to the progression of the patient's symptoms and extensive nature of the tumor on imaging, the patient was taken to the operating room for left medial maxillectomy, left total ethmoidectomy, left sphenoidotomy, left frontal sinusotomy, left orbital decompression, and resection of the left extradural anterior skull base. Intraoperatively, the entire left maxillary sinus was blocked by tumor, causing right sided septal deviation. Upon debulking this area, multiple blood vessels, believed to be branches of the sphenopalatine artery, were noted inside the tumor without appreciable invasion. The medial and inferior walls of the orbit and the anterior portion of the lamina papyracea were involved by tumor and carefully debulked. Exposure of the orbital fat through the floor of the orbit allowed for ocular pressure release. The Neurosurgery team then assisted in the removal of the tumor at the anterior skull base and cribriform plate. Finally, tumor was removed from the frontal recess and frontal sinus anteriorly. Cerebrospinal fluid leak was not observed at the conclusion of the procedure.
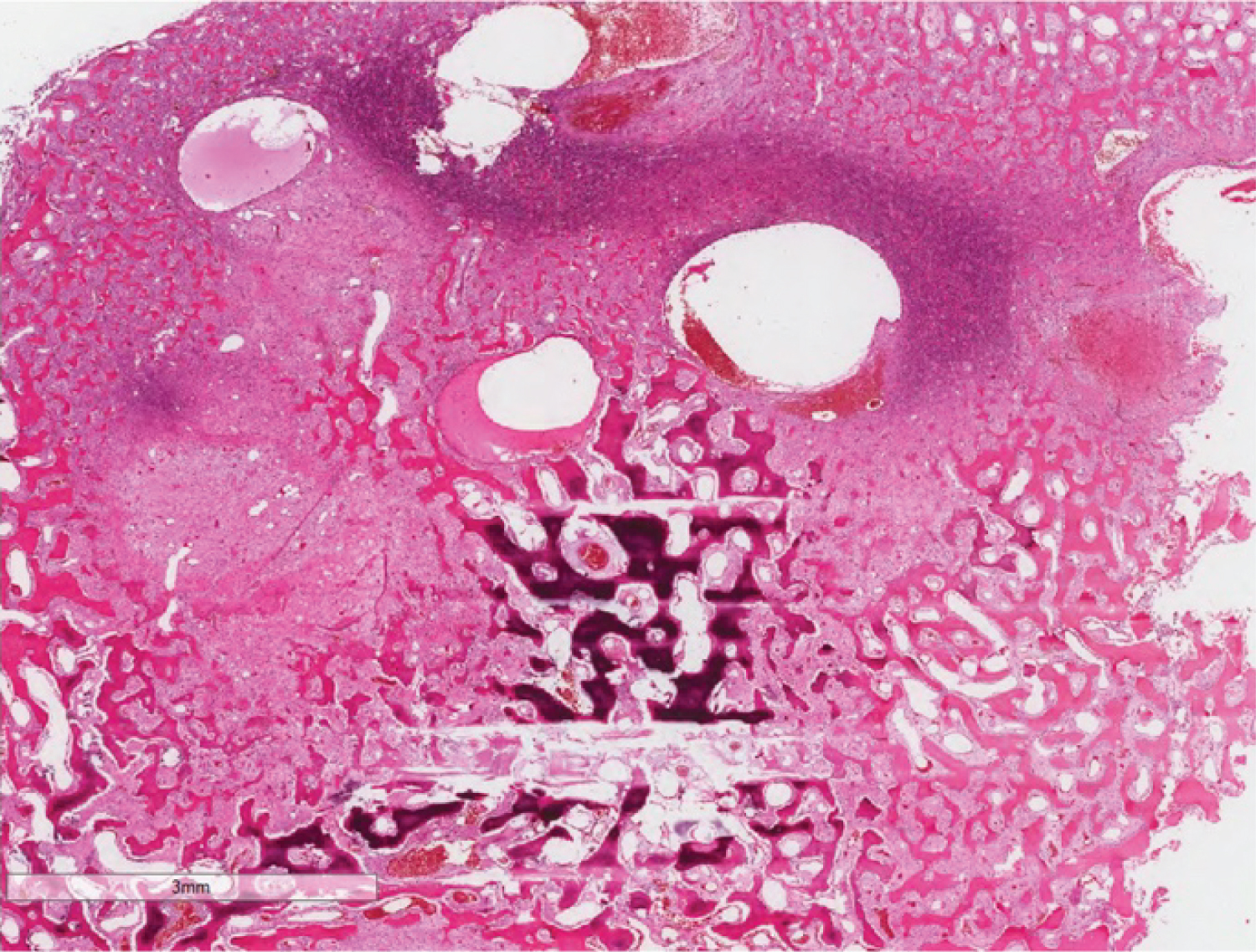
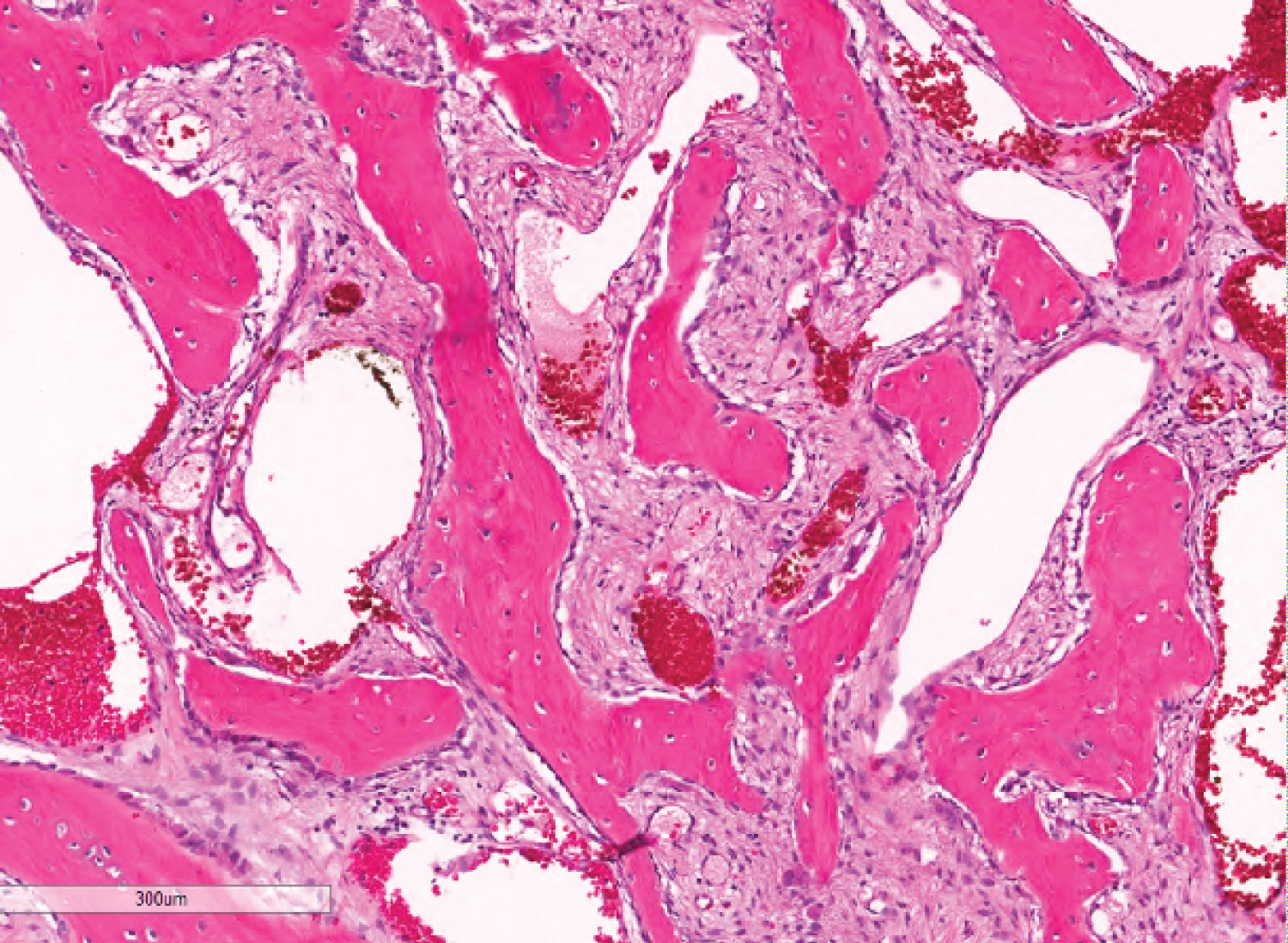
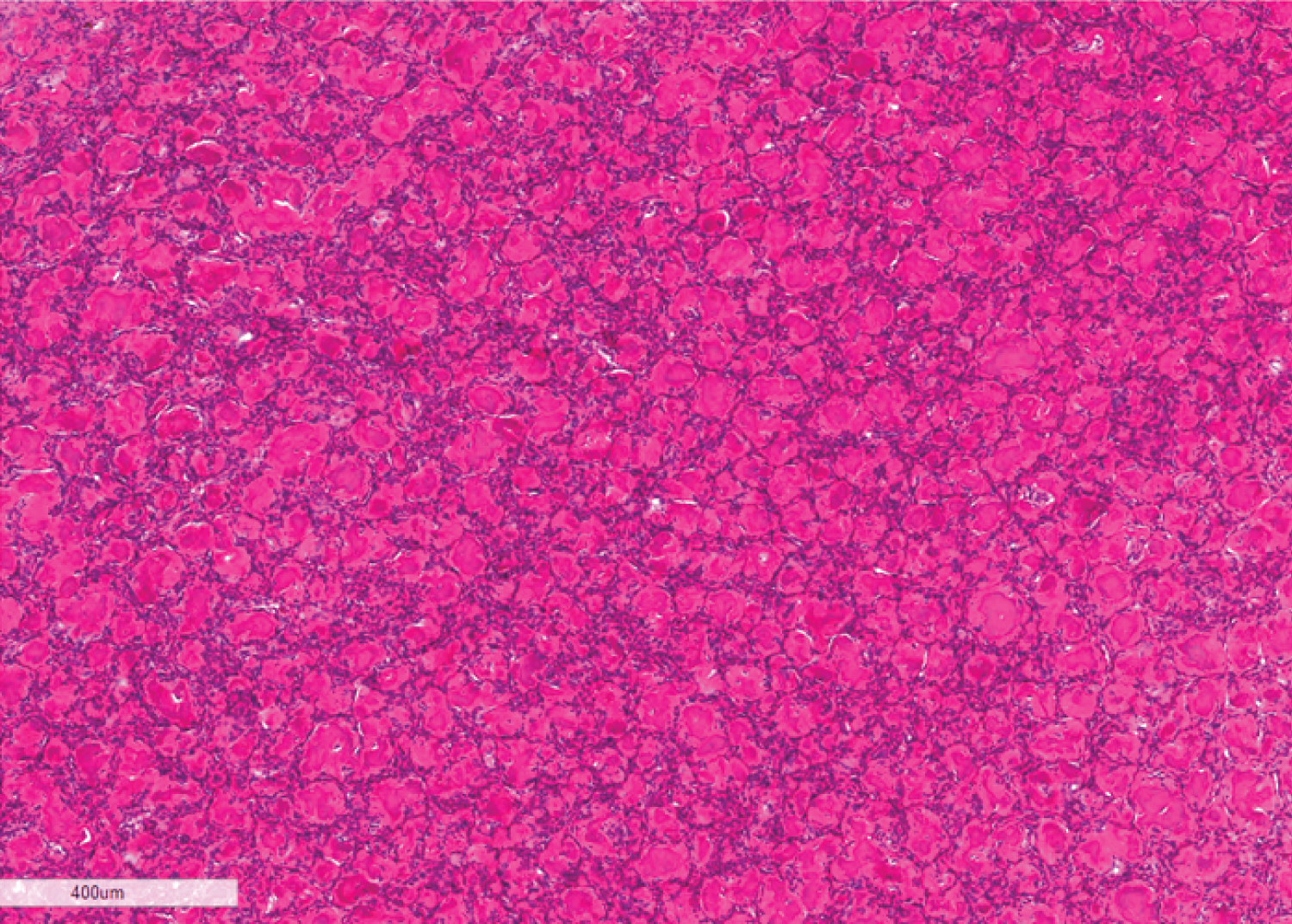
Histologic examination revealed sinonasal mucosa infiltrated by a benign fibro-osseous neoplasm with foci of necrosis. The fibroblastic elements were composed of fasciculated spindle cell proliferations consisting of fibroblast-like cells with uniform elongated, deeply-basophilic nuclei without atypia or mitosis. This framework supported numerous, closely packed, basophilic psammomatous calcifications with eosinophilic rings interspersed with and surrounded by cellular bone trabeculae arranged in an anastomosing trabecular pattern, many of which were rimmed by prominent osteoblasts. In some areas, variably-sized cyst-like spaces containing pale basophilic fluid and blood, consistent with cystic changes, were noted. In other areas, foci of necrosis intermixed with blood and hemosiderin pigment deposits were noted. The neoplastic proliferation was seen in the form of a lobulated growth pattern interspersed by benign sinonasal mucosa. Neither vascular invasion nor tumor emboli were appreciated (Figure 3, Figure 4 and Figure 5). Thus, a final diagnosis of juvenile aggressive ossifying fibroma, psammomatoid variant was made.
Post-operatively, the patient experienced continued left-sided headache, left peri-orbital pain, and persistent left nasal rhinorrhea. Fluid from the left nasal cavity was collected and analyzed for beta-2 transferrin to rule out a potential cerebrospinal fluid leak. The results were reported as negative; thus, the patient was instructed to follow up with an additional sample of nasal fluid if her rhinorrhea persisted.
The patient's case was discussed at the multidisciplinary head and neck tumor board where all available historical data, radiology, pathology, and physical exam findings were reviewed. Post-operative follow-up in the form of regular interval imaging and consideration of an anterior cranio-facial resection, if tumor burden is significantly high, were recommended.
Discussion
Ossifying fibromas are subdivided into cemento-ossifying fibroma (COF) and juvenile ossifying fibroma (JOF) dependent upon the presence of specific histologic features. Unfortunately, ossifying fibroma (OF), JAOF, and fibrous dysplasia (FD) may show significant histological overlap, particularly on small biopsies. The clinical and surgical management of these entities are markedly different; hence it is imperative that these lesions are correctly diagnosed.
As previously stated, ossifying fibromas are benign, slow growing neoplasms that typically present in females in their third and fourth decades of life with most cases occurring in the mandible [4]. On imaging, this entity is a unilocular radiolucent or mixed radiolucent and radiopaque lesions with well-demarcated borders. On gross examination, it is yellow-tan and gritty. Hypercellular fibroblastic stroma without atypia or mitosis, in addition to a quilted pattern of osteoid or bone, is typically seen on histologic examination. Treatment may range from simple enucleation to surgical excision, depending on the extent of the lesion. Recurrence is rare. Of note, multiple ossifying fibromas are associated with hyperparathyroidism-jaw tumor (HJT) syndrome, a hereditary disorder presenting with hyperparathyroidism by way of parathyroid adenoma or carcinoma, and rarely with renal cysts or Wilms' tumors. Identification of this lesion is important as HJT is associated with development of other epithelial tumors, including pancreatic adenocarcinoma, papillary renal cell carcinoma, and testicular mixed germ cell tumor. Mutations in HRPT2, a tumor suppressor gene which encodes the parafibromin protein, have been identified in patients with HJT syndrome [5].
Juvenile aggressive ossifying fibroma (JAOF) is a rare, benign fibro-osseous tumors characterized by rapid growth involving the paranasal sinuses and craniofacial bones in 85% of patients. The calvarium is involved in approximately 12% of cases. In jaw lesions, COF is considered to be an odontogenic tumor and is thought to arise from mesenchymal cells of the periodontal ligament, which form cementum, osteoid, and fibrous tissue [2]. By contrast, JAOF is of uncertain origin, and is generally believed to originate from osseous mesenchyme, as most of these tumors affect sites distant from the odontogenic precursor tissue of COF [1,2,6]. JAOF is a neoplasm of childhood with an equal gender distribution. According to some authors, it has a slight male predilection [7,8]. Clinically JAOF presents with facial asymmetry, facial pain, and chronic sinusitis due to rapid growth and obstruction. Radiological findings include a well circumscribed, radiolucent, uni- or multi-lobulated lesion with varying opacification depending on calcification and cystic areas. CT scans may reveal mixed soft tissue and bone densities with peripheral thinning of the affected bone [7,9]. Grossly, cut surfaces may display curvilinear hemorrhagic strands which are unique to JAOF.
Two pathological variants of JAOF have been identified: Trabecular and psammomatoid. The trabecular variant commonly involves the mandible and occurs in patients less than 12-years-old. Histologically, the stroma is hypercellular and composed of fibroblastic spindle cells with long and slender osteoid forming "paint brush strokes" together with scattered giant cells. In contrast, the psammomatoid variant mostly involves the paranasal sinuses with the ethmoid, frontal, maxillary, sphenoid, and temporal bones being affected, respectively [4,6,10,11]. Cases have been reported in patients from 3-months-old to 72-years-old with the average ranging from 16-33 years-old [3,4,6,11]. This variant shows hypercellular fibroblastic stroma containing numerous closely packed spherical basophilic ossicles - resembling psammoma bodies surrounded by eosinophilic rings - along with prominent osteoblastic rimming. Cystic degeneration and aneurysmal bone cyst formation have been reported in both variants [3,12].
The pathologic features of the current case are a classic example of psammomatoid variant of JAOF, which exhibited cystic changes and foci of necrosis. This is thought to be due to local vascular impairment which leads to foci of ischemic necrosis characterized by pale eosinophilic acellular zones with hemorrhagic and hemosiderin deposition. Clinically, the patient experienced significant facial deformity accompanied by neurologic manifestations - including headaches, anosmia, and blurred vision - likely due to cranial compression by the tumor.
The etiopathogenesis for JAOF is poorly understood, but nonrandom breakdown at Xq26 and 2q33 resulting in (X;2) translocation has been reported in a small number of orbital "cemento-ossifying fibromas" [13]. A recent study performed by Tabareau-Delalande, et al. [14], revealed that JAOFs with long arm rearrangements covering the MDM2 and RASAL1 genes on chromosome 12 may represent a possible marker for the more extensive and aggressive forms of this entity. In addition, MDM2 amplification via FISH was noted in 70% of cases without concordant IHC over expression [14]. GNAS mutation, associated with fibrous dysplasia, has not been found in tested JAOF tumors.
Treatment for JAOF ranges from enucleation and curettage to resection of the tumor with clear margins [15,16]. Small lesions are treated with curettage or ostectomy, whereas extensive and deep lesions with infiltrating borders extending into the base of the skull may be treated with complete endoscopic resection or even craniotomy. Many authors have reported a high recurrence rate after conservative or minimally-invasive treatment (30-58% of cases); thus, complete surgical resection remains the preferred line of treatment and portends an excellent prognosis [7,8,17,18].
Pertinent differential diagnoses to consider include fibrous dysplasia, cementoblastoma, osteosarcoma, osteoblastoma, and aneurysmal bone cyst.
Fibrous dysplasia (FD) typically has irregularly shaped and poorly organized woven bone, without osteoblastic rimming, surrounded by hypocellular stroma. This entity is caused by a missense mutation in the GNAS gene, which drives the formation of abnormal bone [3,4]. Children and adolescents are most affected by this entity, with the maxilla and femur being the most common sites. Radiographically, this lesion is radiolucent with indistinct borders and has a "ground glass" appearance [3]. Polyostotic fibrous dysplasia may be associated with endocrinopathies such as McCune-Albright Syndrome. Nonetheless, treatment typically consists of watchful waiting and delayed surgical intervention as these lesions tend to stabilize with skeletal maturation [3].
Cementoblastoma is a dense, slow-growing mass associated with the tooth root. These lesions arise from the periodontal ligament region - with irregular lacunae, plump cementoblasts, and prominent basophilic reversal lines. Radiographically, loss of the tooth root outline and obliteration of the periodontal ligament space is common. Recurrence is rare with complete excision of the lesion [3].
Osteosarcoma may be ruled out based on its characteristic radiographic appearance of bony destruction and periosteal reaction. Histologically, malignant cells with nuclear pleomorphism, atypical mitoses, and delicate strands of osteoid are appreciated. Surgical excision with clear margins is the treatment of choice. Subsequent chemotherapy may be warranted, especially for high grade lesions; however, radiation therapy is not necessary as this entity is radioresistant. Local recurrence is reported in up to 25% of cases [3,19].
Osteoblastoma shows remodeling of newly formed, thick, bony trabeculae with sheets of epithelioid osteoblasts and trabecular rimming. Features of pleomorphism or atypical mitosis are uncommon. Radiographic imaging will show a sharply circumscribed, lytic lesion with varying degrees of mineralization. Most cases do not recur after curettage and/or local excision [3].
Aneurysmal bone cyst (ABC) presents as cystic spaces filled with blood separated by fibrous septa alternating with solid areas supporting osteoclast-like giant cells. The cystic spaces and septa are lined by fibroblasts, myofibroblasts, and histiocytes but not endothelium [15,16,20-22]. These lesions are radiolucent, uni- or multilocular, and with well-defined borders. Up to 10% can have a mixed radiolucent/radiopaque pattern. Curettage with or without embolization are the treatment of choice [3,23]. Recurrence is typically seen in lesions that extend into the soft tissue. Additionally, ABC may occur in fibrous dysplasia, ossifying fibromas, and other bone lesions. ABC is also highly associated with recurrent JAOF.
Conclusion
Juvenile aggressive ossifying fibromas are rare, benign, but locally aggressive fibro-osseous lesions. Diagnosis requires clinical, radiologic, and pathologic correlation. It is imperative to rule out other fibro-osseous lesions and arrive at the appropriate diagnosis as management and prognosis may differ significantly. Recurrence and corresponding complications may develop in up to 50% of patients, therefore complete excision is critical. Follow-up imaging is recommended for those in which complete excision is not practical.
References
- Eversole R, Su L, El-Mofty S (2008) Benign fibro-osseous lesions of the craniofacial complex. A review. Head Neck Pathol 2: 177-202.
- Animasahun BA, Kayode-Awe G, Kusimo OY (2019) Juvenile ossifying fibroma in a Nigerian boy: A rare case report. AME Case Rep 3: 20.
- El-Naggar AK, Chan JKC, Grandis JR, et al. (2017) Odontogenic and maxillofacial bone tumours. WHO Classification of Head and Neck Tumours. Lyon: International Agency for Research on Cancer. 203-260.
- Shi RR, Li XF, Zhang R, et al. (2013) GNAS mutational analysis in differentiating fibrous dysplasia and ossifying fibroma of the jaw. Mod Pathol 26: 1023-1031.
- Haven CJ, Wong FK, Eveline WCM Van Dam, et al. (2000) A genotypic and histopathological study of a large dutch kindred with hyperparathyroidism-jaw tumor syndrome. The Journal of Clinical Endocrinology & Metabolism 85: 1449-1454.
- El-Mofty S (2002) Psammomatoid and trabecular juvenile ossifying fibroma of the craniofacial skeleton: two distinct clinicopathologic entities. Oral Surgery Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics 93: 296-304.
- Nguyen S, Hamel M-A, Chenard-Roy J, et al. (2019) Juvenile psammomatoid ossifying fibroma: A radiolucent lesion to suspect preoperatively. Radiology Case Reports 14: 1014-1020.
- Keles B, Duran M, Uyar Y, et al. (2010) Juvenile Ossifying Fibroma of the Mandible: A Case Report. Journal of Oral and Maxillofacial Research 1: e5.
- Khoury NJ, Naffaa LN, Shabb NS, et al. (2002) Juvenile ossifying fibroma: CT and MR findings. Eur Radiol 12: S109-S113.
- Shekhar MG, Bokhari K (2009) Juvenile aggressive ossifying fibroma of the maxilla. J Indian Soc Pedod Prev Dent 27: 170-174.
- De Noronha Santos Netto J, Machado Cerri J, Miranda AM, et al. (2013) Benign fibro-osseous lesions: Clinicopathologic features from 143 cases diagnosed in an oral diagnosis setting. Oral Surg, Oral Med, Oral Pathol and Oral Radiol 115: e56-e65.
- Peterson BR, Nelson BL (2015) Juvenile active ossifying fibroma. Head and Neck Pathology 9: 384-386.
- Sawyer JR, Tryka AF, Bell JM, et al. (1995) Nonrandom chromosome breakpoints at Xq26 and 2q33 characterize cemento-ossifying fibromas of the orbit. Cancer 76: 1853-1859.
- Tabareau-Delalande F, Collin C, Gomez-Brouchet A, et al. (2015) Chromosome 12 long arm rearrangement covering MDM2 and RASAL1 is associated with aggressive craniofacial juvenile ossifying fibroma and extracranial psammomatoid fibro-osseous lesions. Mod Pathol 28: 48-56.
- Aboujaoude S, Aoun G (2016) Juvenile trabecular ossifying fibroma of the maxilla: A case report. Medical Archives 70: 470-472.
- Rai S, Kaur M, Goel S, et al. (2012) Trabeculae type of juvenile aggressive ossifying fibroma of the maxilla: Report of two cases. Contemp Clin Dent 3: 45-50.
- Zama M, Gallo S, Santecchia L, et al. (2004) Juvenile active ossifying fibroma with massive involvement of the mandible. Plast Reconstr Surg 113: 970-974.
- Maria A, Sharma Y, Malik M (2013) Juvenile ossifying fibroma of mandible: A case report. J Maxillofac Oral Surg 12: 447-450.
- Nelson BL, Phillips BJ (2019) Benign fibro-osseous lesions of the Head and Neck. Head Neck Pathol 13: 466-475.
- Phattarataratip E, Pholjaroen C, Tiranon PA (2013) Clinicopathologic analysis of 207 cases of benign fibro-osseous lesions of the jaws. Int J Surg Pathol 22: 326-333.
- Bohn OL, Kalmar JR, Allen CM, et al. (2010) Trabecular and psammomatoid juvenile ossifying fibroma of the skull base mimicking psammomatoid meningioma. Head and Neck Pathology 5: 71-75.
- Osunde O, Iyogun C, Adebola R (2013) Juvenile aggressive ossifying fibroma of the maxilla: A case report and review of the literature. Ann Med Health Sci Res 3: 288-290.
- Silva CA, Silva AD, Soares JA, et al. (2011) Trabecular juvenile ossifying fibroma with aneurysmal bone cyst: A rare presentation. Pediatr Dent 33: 388-391.
Corresponding Author
Dr. Sravan Kavuri, MD, Department of Pathology, Medical College of Georgia, 1120 15th Street, BF 103B, Augusta GA 30912, USA, Tel: (706)-721-2771
Copyright
© 2020 Ullah A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.