DNA Adaptor Technology as PCR Application
Abstract
PCR (polymerase chain reaction) is a popular analytical method in genetics of organisms. By using gene-specific primers, a small single region in the huge genomic DNA is selectively amplified by PCR. Thus, PCR is quite frequently used for determination of gene alleles and to identify nucleotide sequences of genes. For these variety of analytical purposes, PCR method is often combined with the other DNA-related methods such as sequence-dependent DNA digestion with restriction enzymes, bisulfite reaction of genomic DNA for estimation of cytosine methylation, and usage of adaptor DNA for advanced analyses. This review will focus on the properties of adaptor DNAs, and will introduce how these short synthetic DNAs help analysis of DNA sequences.
Introduction
The principles of PCR (polymerase chain reaction) are well known, and they are written in the textbooks [1]. Nonetheless, the modes of its reactions should be remembered to fully understand the analytical methods using PCR reactions. After DNA melting and primer annealing, DNA is elongated by the nucleotides attached to 3' hydroxyl groups of DNA end. The attached nucleotides are selected so that they are complementary to the other DNA strand. Deoxycytidine (C) and deoxyguanosine (G) are complementary to each other, and thymidine (T) and deoxyadenosine (A) are complementary to each other. C, G, T, and A are often simply called as cytosine, guanine, thymine and adenine for their names of base residues. Uracil (U) base is not ordinally found in DNA but in RNA. U is complementary to A. Primers, typically around 20-bases long oligo DNAs, are used to initiate PCR reactions. Primers are designed so that they are complementary to the target DNA sequences of PCR amplification. There is also clear discrimination between the ordinary type of DNA polymerase (such as Taq DNA polymerase, Takara, Kusatsu, Japan) and high-fidelity type of DNA polymerase (such as KOD DNA polymerase, Toyobo, Osaka, Japan). The latter is preferred in simple PCR analyses and DNA cloning, for its low frequency of mutation during PCR amplification. The double-stranded DNA end is flat (blunt-ended) with the latter type, but an extra A residue is attached to the 3' end and protruding with the former type. On the other hand, the former type must be used when the template DNA includes irregular nucleotides (bases) such as U and inosine (I).
Difference in the phenotypes of organisms are caused by different allelic sequences in the gene. For example, red apples have a different allele of the MdMYB1 gene from the alleles of yellow apples [2]. Alleles can be determined by simple PCR amplification of the gene, if there are insertion/deletion mutations between different alleles. But more frequently, there are not such clear mutations between alleles, and there are simply single nucleotide mutations (differences/polymorphisms). In such cases, DNA regions including single nucleotide polymorphisms are PCR amplified and digested with restriction enzymes. Restriction enzymes usually recognize 6-base DNA and digest (cut) DNA within this sequence. If there is a mutation in the 6-base DNA sequence, the restriction enzyme does not cut DNA. Thus, after electrophoresis of the digested DNA, alleles are determined by checking if the PCR-amplified DNA was cut by the restriction enzyme. Variety of the 6-base target sequences of restriction enzymes, such as 5'-GGATCC-3' (Bam HI), 5'-GAATTC-3' (Eco RI), and 5'-AAGCTT-3' (Hin dIII) enables selection of a suitable restriction enzyme to some extent: A specific 6-base sequence appears in the DNA sequence at approximately once in 4 kb (kilo-bases) on average. This kind of PCR markers, based on digestion with restriction enzymes, are called cleaved amplified polymorphic sequence (CAPS). If the CAPS marker is not even found within the target sequence, derived CAPS (dCAPS) marker is designed, which generates the target sequences of restriction enzymes by introduction of nucleotide base mutations by primer sequences [3]. Otherwise, nucleotide polymorphisms are detected by simple direct sequencing of genomic DNA.
One of the other representative PCR-based analytical method of genomic DNA is bisulfite sequencing. Cytosine (C) residues are converted to 5-methylcytosine in some cases in the living cell, then it causes changes in gene expressions (activities). Bisulfite reaction of genomic DNA is utilized to determine the methylation status. Bisulfite converts C residues to U, but does not convert 5-methylcytosine. The bisulfite-treated DNA is amplified by using gene-specific primer, and its nucleotide sequence is analyzed after cloning into plasmids. In the DNA sequences, methylated C remains as C, but non-methylated C residues are changed to T, after PCR amplification. The difference in DNA sequence enables measurement of C methylations. Here, bisulfite-treated genomic DNA is digested with the ordinary-type DNA polymerase. There are also specially prepared DNA polymerases for bisulfite sequencing analysis, such as KOD Multi & Epi (Toyobo) and EpiTaq HS (Takara). These two enzymes have similar activities, but only the former enzyme succeeds to amplify target DNA under certain combinations of template DNA and primers, or vice versa.
Methods using DNA adaptors are another good examples of the application of PCR technologies. Thus, the analytical methods using DNA adaptors will be demonstrated in this review. The most frequently used adaptor-related technology will be genome walking (also called 'chromosome walking'), but the DNA adaptors are also used for DNA marker analyses such as dCAPS and direct sequencing approach. PCR amplification of tiny amounts of genomic DNA will be another nice technique with DNA adaptor.
A Popular Adaptor DNA Sequence for Genome Walking
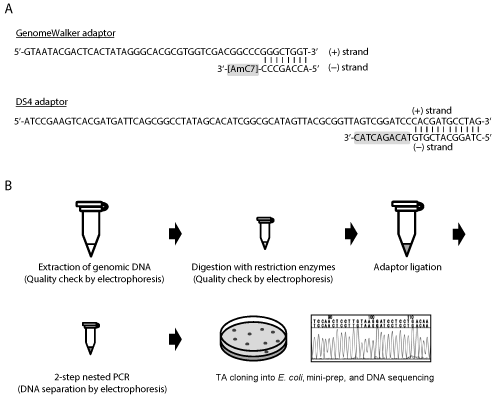
Figure 1A shows the structure of popular DNA adaptor. This adaptor will be called 'Genome Walker adaptor' in this review, because this adaptor sequence is used in the Genome Walker kit released from Clontech/Takara. The Genome Walker adaptor consists of 48-base (+) strand (5'-GTA ATA CGA CTC ACT ATA GGG CAC GCG TGG TCG ACG GCC CGG GCT GGT-3') and 8-base (-) strand (5'-ACC AGC CC-AmC7-3'). These DNA sequences are complementary to each other, then they form partly double-stranded DNA upon mixture in the solution. The 3' end of the (-) strand is modified (attached) with a C7 amine 'amino modifier C7' (AmC7) to inhibit the formation of fully double-stranded adaptor sequence during PCR analysis. If the 3' end of the (-) strand is not modified, there will be non-specific amplification of any DNA sequence in PCR. The right end of this adaptor is blunt-ended (flat), then the adaptor can be attached to blunt-ended DNA ends, such as genomic DNA digested with the restriction enzyme Dra I, Eco RV, Pvu II, Sca I, or Ssp I. When the restriction enzyme forms cohesive ends at the end of digested DNA, the adaptor sequences should be modified so that the right end fits the structure of the digested DNA. Cohesive-ended DNA needs preparation of separate DNA adaptors, but analysis with cohesive-ended DNA seems to be more successful than with blunt-ended DNA based on our experience and also according to the cases in plasmid ligations [4], probably because the DNA fragments are more tightly attached to the adaptor. Genomic DNA is also often methylated, so genomic DNA had better be digested with methylation-insensitive restriction enzymes.
The scheme of adaptor-PCR analysis is shown in the Figure 1B. After digestion of genomic DNA with several (4 to 8) restriction enzymes separately, DNA adaptors which had been prepared for each restriction enzyme are attached to genomic DNA by ligation reaction. The adaptor-ligated genomic DNA fragments are then PCR amplified with adaptor primers and gene-specific primers. The PCR is performed as a two-step nested PCR. This reaction will amplify down-stream genomic DNA sequences of the gene-specific primers. A typical application of this adaptor-PCR technology is the isolation of gene promoters. The 5'-UTR (untranslated region) and 1st exon sequences are first prepared by RT (reverse transcription)-PCR analysis, then these sequences are used to design gene-specific primers in the reverse orientation. Adaptor PCR using these primers will amplify upstream (promoter) sequence of the target gene. Please refer to former publications for details of adaptor-PCR protocols [5-8]. The Genome Wakler adaptor seems to have been used in most of the past adaptor-PCR studies [5-9].
DS4 Adaptor for Genome Walking
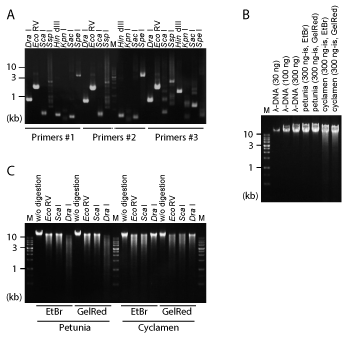
A trial was also made to improve the performance of the adaptor. In our previous report [10], we designed a new DNA adaptor named 'DS4 adaptor'. The structure of the DS4 adaptor is also shown in Figure 1A. DS4 adaptor is the combination of a 79-base (+) strand (5'-ATC CGA AGT CAC GAT G ATT CAG CGG C CTA TAG CAC ATC GGC GCA TAG TTA CGC GGT TAG TCG GAT CCC ACG ATG CCT AG-3') and a 22-base (-) strand (5'-CTA GGC ATC GTG TAC AGA CTA C-3'). Unlike the Genome Walker adaptor, the 3' end of the (-) strand is not chemically modified. But instead of chemical modification, the 10 nucleotides at the 3' end of this oligo DNA is not complementary to the (+) strand of the DS4 adaptor. Attachment of this 10-base mismatch sequence to the (-) strand is enough for inhibition of the amplification of non-target DNA sequence, and the adaptor PCR analysis using the DS4 adaptor successfully amplifies long, unknown franking sequences of the target genes (Figure 2A) [10,11].
Another advantage of the DS4 adaptor lies in the sequence of the (+) strand. A high rate of GC content in Genome Walker adaptor was reduced in DS4 adaptor, and the adaptor was also elongated in DS4. These changes merit stable amplification of the target sequence by PCR analysis. In other words, longer 'adaptor primers' such as DS4-AP1 (5'-GAA GTC ACG ATG ATT CAG CGG CCT ATA GCA-3') and DS4-AP2 (5'-GGC CTA TAG CAC ATC GGC GCA TAG TTA CG-3') are designed for DS4, allowing PCR amplifications at high annealing temperature by using high-fidelity DNA polymerase. This modification increased the possibility of successful PCR amplification of the target sequence. DS4 adaptor sequence is based on a random nucleotide sequence generated by myself, and the candidate nucleotide sequences were also screened so that they are suitable for primer design, by using the computer program 'Primer3' [12,13]. The DS4 sequence was then chosen so that it is not similar to any known DNA sequence of any organism. Thus, the DS4 sequence does not match any sequence in the BLAST search at the website of NCBI [14], further ensuring specific DNA amplification.
Adaptor Attachment to dCAPS and Degenerate Primers
Application of DNA adaptors is not restricted to genome walking. For example, DS4 adaptor was used to improve the performance of PCR primers of DNA markers. There is usually just around 20-bp difference in the length of DNA fragments, when different alleles are detected by dCAPS markers. Such small difference in DNA size is not always discriminated by agarose gel electrophoresis. Such disadvantage of dCAPS primers derives from the structure of these primers, and seems to have been inevitable until recently. In our recent report, however, the difference in DNA fragments was successfully elongated to nearly 100 bp [15]. This was done by simply attaching the DS4 sequence to the dCAPS primer of apple MdMYB1 gene (Figure 3A). The resultant DNA fragments were consistent to those obtained with the original primer set, and the DNA bands were more clearly separated from each other. In this case of the improvement of dCAPS primer, DS4 sequence was used only to elongate the PCR-amplified DNA fragment. Nevertheless, this method potentially improves the performance of any reported or newly designed dCAPS primers. In the case of the dCAPS primer for MdMYB1 gene, DS4 sequence in the forward primer did not inhibit PCR amplification, and on the contrary, it looked to improve PCR amplification from apple genomic DNA.
DS4 adaptor was also used to generate a unique PCR marker for determination of apple S alleles [16]. Here, 14 different alleles of apple S-RNase genes are amplified by degenerate primers. The forward degenerate primer is also fused with DS4 at the 5' end (Figure 3A). This set of adaptor-fused degenerate primers was named APPLid (apple S-allele identifier). APPLid primers amplified genomic sequence of apple S-RNase from any tested cultivars. The DS4 sequence attached to the forward primer enables direct sequencing analysis of the amplified DNA, then both two S alleles of diploid apple cultivars are determined from the nucleotide sequences (Figure 3B). This strategy of high-throughput and sequencing-based DNA marker with DS4 adaptor will be also applicable to many other organisms.
Further Application of DNA Adaptors
Adaptor ligation is also utilized for whole-genome amplification (WGA) [17]. This method is used for even (non-biased) amplification of small amount of whole genome extracted from biological samples. The procedure consists of shearing genomic DNA in hydrodynamic shearing machine to 0.5-2.0 kb, digestion with BAL31 nuclease, treatment with T4 DNA polymerase for end-filling, adaptor ligation, and PCR amplification. WGA by this protocol (called PRSG) successfully amplifies more than 99% of the whole genome when high-quality DNA is used as the template, but this figure drops to 50-70% when low-quality DNA is used [18].
Another possible application of the DNA adaptor would be direct sequencing in bisulfite-PCR analysis. For analysis of DNA methylation states, bisulfite-treated genomic DNA is usually PCR amplified and cloned into plasmid vectors. This method needs multiple steps, then time and labor are consumed. If bisulfite-treated genomic DNA is analyzed by direct sequencing by using adaptor-fused primer, it will provide rapid estimation of the methylation state. We actually made this trial, but not successful yet.
High-Quality DNA Extraction and Purification for Successful PCR Analyses
It may not be widely recognized, but a critical factor affecting the success rate of adaptor-assisted PCR analyses is the quality and quantity of DNA extracted from sample tissues, according to our experience. Here, I will introduce the best protocols to obtain high-density genomic DNA at high purity. These protocols are successful in virtually all plant species. The protocols are not tested for bacteria and animals, but may also provide good hints for DNA preparation from these organisms.
The difficulty of DNA extraction is quite variable between plant species. The 'alkaline PVPP buffer' may be the only method that can extract high-density DNA from any plants species [19,20]. This DNA extraction buffer contains polyvinylpolypyrrolidone (PVPP) and high concentration (4 M) of NaCl. These unique constituents help DNA extraction from well-homogenized leaf tissues when heated at 60 ℃ for 30 min. The purity of crude DNA solution is quite low. Then, the actual DNA concentration cannot be determined by measurement of UV absorbance at 260 nm. Purity of DNA is estimated by gel electrophoresis of extracted DNA and compared with standard pure DNA such as λ-DNA. That is, the DNA amount estimated as 300 ng-is is electrophoresed together with 300 ng λ-DNA. 'ng-is' represents 'ng in spectrometry' here (Figure 2B). The density of crude DNA bands observed after agarose gel electrophoresis is almost always much lower than λ-DNA, indicating that the purity of crude DNA is quite low. In addition, crude DNA solution often contains RNAs, which are observed as small ladder bands of ribosomal RNAs after electrophoresis. The amount of DNA included in solution is also estimated by electrophoresis of the solution equivalent to 10-mg fresh weight of leaf sample, together with λ-DNA. The DNA yield from plant leaves is usually greater than 100 ng per 10-mg fresh weight with alkaline PVPP buffer. Such high level of efficiency in DNA extraction is necessary to obtain enough amounts of DNA samples for adaptor PCR analyses.
DNA purification also matters. DNA purity will have influence on PCR amplification. In addition, DNA must be purified to be properly cut by restriction enzymes: Impurity in DNA solution inhibits the enzymatic reaction. Similarly, more highly purified DNA should be prepared when DNA methylation state is examined by bisulfite-PCR analysis. When the target plant species is relatively easy for DNA extraction, DNeasy plant mini kit (QIAGEN, Venlo, Netherlands) would be the best choice for routine preparation of DNA samples. Otherwise, when DNA is extracted from recalcitrant plants such as rose and cyclamen, DNA is extracted by using alkaline PVPP buffer and purified by ultracentrifugation in CsCl solution [19]. Purified DNA is properly digested by restriction enzymes, as confirmed by electrophoresis in Figure 2C. Recently, I also noticed that 'silica monofas' column of the DNA purification/extraction kit MonoFas (GL Sciences, Tokyo, Japan) is also effective on genomic DNA purification within quite short time. This kit may become the standard protocol of genomic DNA purification in the near future.
Conclusion
DNA adaptors add unique applications to PCR methods, and have greatly assisted advances in genetic analyses and biotechnology, such as characterization of T-DNA tagged lines [21] and promoter isolation [9,11]. T-DNA tagged lines, especially T-DNA tagged mutants of Arabidopsis thaliana released from Salk Institute [22] have greatly contributed to the progress of plant genetics. Promoters isolated from horticultural plants by adaptor-PCR method also contribute to transgenic modification of ornamental traits [9,11]. Even better adaptor-related techniques such as DS4 adaptor and adaptor-fused DNA markers were devised recently [10,15,16]. Together with the advances in the protocols for preparation of pure genomic DNA, DNA adaptors are providing much more applications to genetic studies.
References
- Sambrook J, Russell DW (2001) The Basic Polymerase Chain Reaction. In: Molecular Cloning. Cold Spring Harbor Laboratory Press, New York, 8.18-8.24.
- Takos AM, Jaffe FW, Jacob SR, et al. (2006) Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol 142: 1216-1232.
- Neff MM, Turk E, Kalishman M (2002) Web-based primer design for single nucleotide polymorphism analysis. Trends Genet 18: 613-615.
- Sambrook J, Russell DW (2001) Cloning in Plasmid Vectors. In: Molecular Cloning. Cold Spring Harbor Laboratory Press, New York, 1.19-1.24.
- Siebert PD, Chenchik A, Kellogg DE, et al. (1995) An improved PCR method for walking in uncloned genomic DNA. Nucl Acid Res 23: 1087-1088.
- Yamamoto YY, Tsuhara Y, Gohda K, et al. (2003) Gene trapping of the Arabidopsis genome with a firefly luciferase reporter. Plant J 35: 273-283.
- Ichikawa T, Nakazawa M, Kawashima M, et al. (2003) Sequence database of 1172 T-DNA insertion sites in Arabidopsis activation-tagging lines that showed phenotypes in T1 generation. Plant J 36: 421-429.
- Kasajima I, Yoshizami T, Ichikawa T, et al. (2010) Possible involvement of ploidy in tolerance to boron deficiency in Arabidopsis thaliana. Plant Biotechnol 27: 435-445.
- Sasaki K, Yamaguchi H, Kasajima I, et al. (2016) Generation of novel floral traits using a combination of floral organ-specific promoters and a chimeric repressor in Torenia fournieri Lind. Plant Cell Physiol 57: 1319-1331.
- Kasajima I, Ohtsubo N, Sasaki K (2014) Faster, safer, and better DNA purification by ultracentrifugation using GelRed stain and development of mismatch oligo DNA for genome walking. Biosci Biotechnol Biochem 78: 1902-1905.
- Kasajima I, Ohtsubo N, Sasaki K (2017) Combination of Cyclamen persicum Mill. floral gene promoters and chimeric repressors for the modification of ornamental traits in Torenia fournieri Lind. Hortic Res 4: 17008.
- Untergasser A, Cutcutache I, Koressaar T, et al. (2012) Primer3 - new capabilities and interfaces. Nucl Acid Res 40: e115.
- Koressaar T, Remm M (2007) Enhancements and modifications of primer design program Primer3. Bioinformatics 23: 1289-1291.
- https://blast.ncbi.nlm.nih.gov/Blast.cgi.
- Kikuchi T, Kasajima I, Morita M, et al. (2017) Practical DNA markers to estimate apple (Malus × domestica Borkh.) skin color, ethylene production and pathogen resistance. J Hortic 4: 211.
- Kasajima I, Kikuchi T, Yoshikawa N (2017) Rapid identification of apple (Malus × domestica Borkh.) S alleles using sequencing-based DNA marker APPLid. Plant Biotechnol 34: 97-106.
- Arneson N, Hughes S, Houlston R, et al. (2008) Whole-genome amplification by adaptor-ligation PCR of randomly sheared genomic DNA (PRSG). CSH Protoc.
- Tanabe C, Aoyagi K, Sakiyama T, et al. (2003) Evaluation of a whole-genome amplification method based on adaptor-ligation PCR of randomly sheared genomic DNA. Genes Chromosomes Cancer 38: 168-176.
- Kasajima I, Sasaki K, Tanaka Y, et al. (2013) Large-scale extraction of pure DNA from mature leaves of Cyclamen persicum Mill. and other recalcitrant plants with alkaline polyvinylpolypyrrolidone (PVPP). Sci Hortic 164: 65-72.
- Kasajima I (2017) Protocol for DNA extraction from any plant species (alkaline PVPP method). Protoc Exch.
- Alonso JM, Stepanova AN, Leisse TJ, et al. (2003) Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301: 653-657.
- SIGnAL (Salk Institute Genomic Analysis Laboratory).
Corresponding Author
Ichiro Kasajima, Faculty of Agriculture, Iwate University, Ueda 3-18-8, Morioka, Iwate 020-8550, Japan, Tel: +81-19-621-6103 (ext) 5225.
Copyright
© 2018 Kasajima I. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.