Using the pLexsy-sat2 Expression System to Investigate Secreted Acid Phosphatases (Saps) in Leishmania Tarentolae
Abstract
Leishmaniasis is a parasitic disease caused by the protozoa Leishmania that affects both humans and animals. This disease is associated mostly with developing countries, especially in warmer climate regions. There are few good therapeutic drugs, so it is necessary to develop more affordable treatments for those stricken by the disease. Leishmania produce Secreted Acid Phosphatases (SAPs) during their growth cycle as an important part of their cell targeting system against host macrophage cells thus allowing for successful infection. Inhibition of these SAPs can be the focus of new drug therapy. Recombinant DNA technology was employed to characterize these enzymes. Expression of two Secreted Acid Phosphatases, from L. major, SAP1 and SAP2, was attempted in Arctic Express E. coli cells using pET45b or Leishmania tarentolae using the pLexsy-sat2 expression system. The prokaryotic systems were carried out first due to ease of expression control. The eukaryotic expression system was used to overcome expression and activity problems experienced with the prokaryotic systems.
Keywords
Leishmania, Secreted acid phosphatases, Enzyme purification, Recombinant DNA, Arctic express cells, pLexsy-sat2 expression system
Introduction
Leishmaniasis is a growing worldwide health concern, especially in developing nations where this disease is often endemic. Human and animal leishmaniasis is caused by some 20 species of Leishmania that are transmitted by sandflies [1]. Although leishmaniasis afflicts about 2 million people annually and threatens some 350 million people in some 90 countries worldwide, the Centers for Disease Control and Prevention consider leishmaniasis a neglected disease [2,3]. The status quo of treatment for the three major forms of leishmaniasis: cutaneous, mucosal, and visceral, is troubling. As reviewed by Pathak and Batra [4], current drugs, such as pentavalent antimonials, amphotericin B, fluconazole, pentamidine, and miltefosine, have serious side effects, some delivery problems, and drug resistance is also being reported. Additionally, the mechanisms of drug action are not always well understood, especially for natural product extracts [4]. We have recently reported that some selected vanadium complexes, such as decavanadate, have in vitro inhibitory effects on growth of Leishmania tarentolae as well as on some enzymes, involved in intermediary metabolism [5]. Since it is well established that orthovanadate (VO4-3) and other vanadium complexes are effective inhibitors of phosphatases [6], this is of interest because Vannier-Santos, et al. [7] have reported that Leishmania secretes Acid Phosphatases (SAP), which have an important role in the infectivity by Leishmania. We have previously evaluated the in vitro effects of the vanadium complexes on the Secreted Acid Phosphatases [8] using partially purified enzyme following the method of Ilg, et al. [9]. We were able to show inhibitory effects by vanadium complexes. However, there are at least two isoforms of Secreted Acid Phosphatase, which differ by molecular weight and degree of glycosylation [9,10,11]. The presence of these isoforms therefore complicated the kinetic evaluation of the inhibition data and we proposed that the expression of recombinant SAPs would be a more useful approach to study their inhibition by vanadium complexes. However, we wanted to study the secreted enzymes from the human parasites, such as L major, since the effects of test compounds on the human parasites is of direct clinical interest. We did not, however, want to have the expense and problems associated with growing these human parasites for direct isolation of their enzymes. Here we report the use of two different expression systems to investigate recombinant L. major Secreted Acid Phosphatases (SAPs).
For recombinant protein production, we evaluated the E. coli expression system first due to the ease of recombinant expression control followed by use of the eukaryotic system. The prokaryotic system (E. coli Arctic Express cells) resulted in no active enzyme, although recombinant protein was evidenced by electrophoresis. The eukaryotic Leishmania cell expression system for recombinant Secreted Acid Phosphatase 1 (SAP1) and Secreted Acid Phosphatase 2 (SAP2) was employed in these studies with the recombinant SAPs since these enzymes are reported to have extensive glycosylation [9,10,11] and we anticipated that glycosylation would increase the possibility of obtaining active enzymes.
Materials and Methods
Cloning of SAP1 and SAP2 genes
Putative secreted acid phosphatase (SAP1 and SAP2) genes are present in the Leishmania major genome (SAP1 accession XM_001687187 and SAP2 accession XM_001687184). The gene sequences are shown in the Supplementary Material Section. Polymerase Chain Reaction (PCR) was used to amplify these genes. For prokaryotic expression, the SAP1 5' primer was 5'-TACTGGATCCCGTGCAGGTGGTACACCGCC-3' and 3' primer 5'-TACTCTCGAGTTAACAATGTCTGCGGTAC-3'. The underlined sequences in the PCR primers indicate a BamHI site (GGATCC) and an XhoI site (CTCGAG) for cloning into pET45b (Novagen). Similarly, the SAP2 gene was amplified using the primers 5'-TACTGGATCCCGTGCAGGTGGTACACCGCC-3' and 5'-TACTCTCGAGTTAAGGCACGCGAGATGTATG-3'. For eukaryotic expression the SAP1 and SAP2 genes were cloned into the PLexsy-sat2 vector (Jena Bioscience) using the BglII and NheI restriction enzyme sites. Amplification primers for SAP1 were 5'-TACAGTAGATCTGCCATGGTGCAGGTGGTGGTACAC-3' and 5'-TACAGTGCTAGCACAATGTCTGCGGTACTC-3'. For SAP2 amplification primer sequences were 5'-TACAGTAGATCTGCCATGGTGCAGGTGGTGGTACAC-3' and 5'-TACAGTGCTAGCAGGCACGCGAGATGTATG-3', with BglII and NheI sites underlined. PCR employed L. major genomic DNA as the template and Taq DNA Polymerase (New England Biolabs). The reaction mixture contained 0.2 mM dNTPs, 4 mM MgSO4, and 1 mM of each primer and were cycled through the PTC-200 Peltier Thermal Cycler thirty times at the following conditions: 94 ℃ for 15 seconds, 50 ℃ for 30 seconds, and 72 ℃ for one minute.
A Purelink™ Quick PCR Purification Kit from Invitrogen was used to purify PCR products. A 0.7% (w/v) agarose gel was used to identify PCR products. Five μL of purified PCR product was electrophoresed (150 V, 35 minutes) with 5 μL of blue orange 6x loading dye from Promega with molecular weight standards (100 bp from Promega). The DNA bands were cut from the gel, isolated using a Purelink™ Quick Gel Extraction Kit from Invitrogen, then restriction enzyme digestion was performed using either BamHI and XhoI (pET45b) or BglII and NheI (pLexsy-sat2) on the purified PCR products and plasmid templates overnight at 37 ℃ following the recommendation of the manufacturer. To confirm that the restriction enzyme digest was successful, the products were evaluated using a 0.7% agarose gel as previously described. The desired samples were then ligated overnight at 4 ℃ into either pET45b or pLexsy-sat2 using T4 DNA ligase (Promega). Expression of the SAP genes using pET45b results in an N-terminal His-tagged protein and expression using pLexsy-sat2 results in C-terminal His-tagged proteins. pET45b confers ampicillin resistance and pLexsy-sat2 contains an NTC resistance gene.
Expression of SAP1 and SAP2 in arctic express E. coli
The recombinant plasmid DNA pET45b-SAP1 and pET45b-SAP2 were transformed into competent Arctic Express E.coli (Arctic Express; DE3CodonPlus RIL E. coli; Agilent Genomics) by incubation on ice for 30 minutes followed by heat shock at 42 ℃ for one minute then being placed on ice again for 2 minutes. After growth at 37 ℃ for one hour cells were plated on LB media with 100 µg/mL ampicillin, 75 µg/mL streptomycin and 34 µg/mL chloramphenicol and grown at 37 ℃ overnight. For recombinant SAP protein expression, a colony was picked from the transformation plates and transferred to a 14 mL sterile tube that contained 5 mL of LB medium supplemented with 50 μg/mL ampicillin, 20 μg/mL gentamycin and 75 μg/mL streptomycin. After 5 hours of incubation, the 5 mL culture was transferred to a sterile flask containing 50 ml of LB supplemented with 50 μg/mL ampicillin, 20 μg/mL gentamycin and 75 μg/mL streptomycin. The 50 mL cultures were grown overnight, transferred to 1 liter LB cultures supplemented with 50 μg/mL ampicillin, 20 μg/mL gentamycin and 75 μg/mL streptomycin, and allowed to grow in a shaker incubator at 37 ℃. When the cultures had an absorbance of 0.8-1.2 at 600 nm, protein expression was induced by the addition of 0.1 g of IPTG. After induction with IPTG, the 1-liter cultures were allowed to grow for 72 hours (3 days) at 11 ℃ in a cold room with stirring.
Expression of SAP1 and SAP2 in Leishmania tarentolae
The pLexsy-sat2-SAP1 and pLexsy-sat2-SAP2 plasmids were cleaved by the SwaI restriction enzyme in a solution of 2 μl NE3 buffer, 1 μl SwaI (New England Biolabs), and 17 μl plasmid DNA. Reaction mixtures were incubated at 37 ℃ overnight (approximately 16 hours). Agarose gel electrophoresis was utilized to confirm completion of the digest and DNA fragments of interest were excised from the gel with a razor blade. The excised DNA was purified using the Purelink™ Quick Gel Extraction Kit from Invitrogen. The DNA samples were stored at -20 ℃ for later use in the transfection of L. tarentolae by electroporation. All work with L. tarentolae was performed under sterile conditions. The DNA samples were electroporated with a Biorad GenePulser Xcell™ at 450 V for 5 msec with concentrated log phase L. tarentolae cells in a 2 mm electrocuvette. The cells were prepared by incubating a 20 mL brain heart infusion (BHI; Becton, Dickinson and Company) medium with 100 μL of L. tarentolae cells (ATCC #30143) following the method of Mendez, et al. [8]. After four days, the cells were pelleted (6,000 xg, 10 minutes, 4 ℃) and resuspended in half of the original volume (10 mL) using fresh medium. The culture was put on ice for ten minutes, electroporated with the given parameters previously described, and immediately returned to ice for an additional 10 minutes. The cells were then transferred into a fresh 10 mL complete BHI culture with hemin (20 µM), penicillin, and streptomycin (10,000 units/mL and 10 mg/mL, respectively; Sigma). After an overnight incubation at 26 ℃, the selective antibiotic NTC (1000X v/v; Jena Bioscience) was added to the cultures to select for the cells that incorporated the gene. A volume of 100 μL of cells was transferred every three days for two weeks into fresh 10 mL BHI media with hemin, pen/strep, and NTC. The SAP1 and SAP2 recombinants were processed and grown separately.
NTC resistance of recombinant strains
Each cell type was transferred to a 10 mL flask containing BHI, hemin, pen/strep, and NTC. The concentration of NTC used in cultures was 1:1000 v/v. The NTC-selective antibiotic resistant cells were evaluated using a standard MTT cell viability procedure [8] at selected times in the growth of SAP1, SAP2, or wild type (non-recombinant) cultures. The results are reported as a mean of n = 4 and error under 5% is not shown.
SAP1 and SAP2 purification
Bacterial cell cultures were harvested by centrifugation at 5000 rpm for five minutes. Cell pellets w in 20 mL lysis buffer (50 mM Tris Base, 100 mM NaCl, pH 7.5), treated with 500 μL protease inhibitors (2 mM AEBSF, 0.3 μM Aprotinin, 130 μM Bestatin, 1 mM EDTA, 14 μM E-64, 1 μM Leupeptin), and broken open in a French Press (Sim-Aminco, Spectronic Instruments; 10,000 psi). Samples were then spun at 15,000 rpm for twenty minutes, and the supernatant was collected for purification on a Ni-NTA AgaroseNi 2+ affinity resin column (Quigen). The column was loaded with resin and equilibrated with lysis buffer before the sample was added to the column. The column was then washed with 50 mL lysis buffer and 50 mL 10 mM Imidazole (in lysis buffer) before elution of bound protein with 10 mL 200 mM Imidazole (in lysis buffer). Imidazole elutions were collected in 1 mL fractions. Protein concentration in each fraction was determined with the Bio-Rad Protein Assay.
For pLexsy expression, SAP1 recombinant cells, SAP2 recombinant cells, or L. tarentolae wild type cells (500 mL) were incubated in a shaking incubator (100 rpm) in the dark at room temperature for 10 days. The cells were pelleted at 9,000 xg, 4 ℃ for 15 minutes. The supernatant was collected and the pH was adjusted to 7 by addition of 67 mM Tris buffer pH 7.4. The cell pellet was resuspended in 50 mL of 67 mM Tris buffer pH 7.4 and lysed using the Sim-Aminco French Press at 10,000 psi. The lysate was centrifuged at 10,000 xg for 30 minutes at 4 ℃ and the supernatant was evaluated for enzyme activity. Ni2+ affinity chromatography and Concanavalin A-Sepharose 4B (Con-A; GE life Sciences) affinity resin were used to isolate these recombinant enzymes. Nickel columns were equilibrated and washed with 67 mM Tris buffer pH 7.4 (20 mL buffer per mL of resin used) and eluted using 67 mM Tris buffer with 200 mM imidazole pH 7.4 (5 mL). The fractions were collected and assayed for acid phosphatase activity. Concanavalin A chromatography was also employed following the manufacturer recommended procedure (Sigma).
Enzyme assays
Using the Ni2+ affinity resin, precolumn, flow through, wash, and eluted fractions from the wild type, SAP1 or SAP2 supernatant were assayed for catalytic activity using the artificial substrate para-nitrophenylphosphate (pNpp, Sigma-Aldrich), which was dissolved in sodium acetate buffer (0.5 M, pH 4.5). These conditions thus select for acid phosphatase activity even though Leishmania do have neutral and alkaline phosphatases. The test samples (450 µL, n = 3) were incubated with the assay buffer and pNPP (final concentration 220 µM) in a final reaction volume of 1.0 mL. The enzymatic reactions were incubated for 36 hours then stopped by the addition of 10 M NaOH (100 µL) after which product formation was evaluated by visible spectroscopy at 405 nm. The correct spectrophotometric blank was used for each buffer. Data are reported as A405nm/36 hours. A protein determination was performed on each fraction using the Bio-Rad reagent and BSA as the standard.
Results
PCR of SAP1 and SAP2
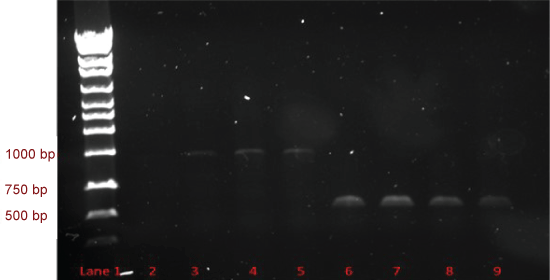
PCR products of SAP1 and SAP2 are shown on an agarose gel in Figure 1. SAP1 products are in lanes 3-5 at approximately 1100 bp, and SAP2 products are in lanes 6-9 at approximately 680 bp. These are the expected sizes for the SAP1 and SAP2 genes.
Chromatography of recombinant protein
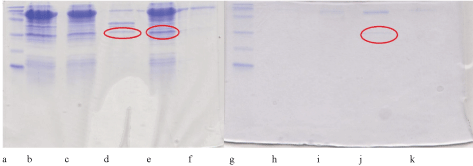
The prokaryotic expression system (E. coli Artic Express cells) was used, but resulted in no active protein (data not shown), although recombinant protein was evidenced by electrophoresis as shown in Figure 2.
NTC resistance of Leishmania recombinant strains
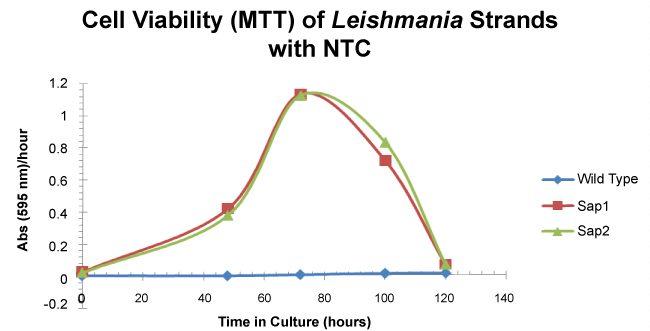
To demonstrate that the pLexsy-sat2 incorporated genes were taken up by L. tarentolae, the cells were grown in complete BHI medium with NTC for 5 days and evaluated by the MTT viability assay. The SAP1 and SAP2 strains are viable for 120 hours in culture reaching the stationary phase at roughly 72 hours. The wild type strain was not viable under the selective antibiotic conditions (Figure 3) over the 5 day culture time.
Enzyme assays using pLexsy-sat2 expression system
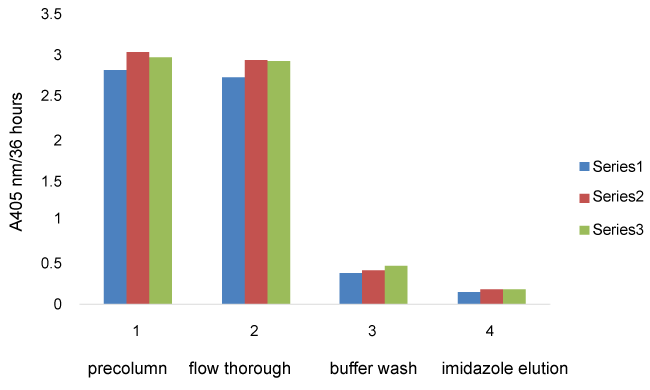
Using lysed cell supernatant, the pre-column fractions from all cell strains showed the highest average absorbance indicating high levels of acid phosphatase activity. The cell flow through and wash fractions (Figure 4) also had very high activity for all 3 cell strains indicating that most SAP activity did not stick to the columns. The activity from SAP1 and SAP2 elution fractions are not significantly different from the wild type elution fraction activity for the French pressed cell supernatants. The same trends were seen with the Con-A purification technique (data not shown). There was not a significant difference in protein concentration for the same fraction from each cell type (data not shown).
Recombinant SAP1 and SAP activity eluding from the column was approximately 6% of the phosphatase activity loaded onto the column whereas approximately 5% of the Secreted Acid Phosphatase activity from the wild type cells loaded onto the column eluded is this same imidazole wash fraction. Thus, it appears that there was no additional recombinant enzyme activity detectable.
Discussions and Conclusions
The successful insertion of the pLexy-sat2 vector containing either the SAP1 or SAP2 gene into Leishmania cells is evidenced by the resistance of these strains to the NTC antibiotic which is not seen in wild type cells. The WT cells were unable to grow in the NTC. An increase in SAP activity is not seen in either the French pressed cells or the cell free supernatants (data not shown) relative to the wild type endogenous activity. Therefore, we conclude that the recombinant enzymes are expressed either at very low levels relative to endogenous enzyme or are not catalytically active. In addition, protein concentration was not found to be significantly different from the SAP1, SAP2, or wild type cell purification techniques. Attempts were made to increase gene expression by altering pH or temperature, but the expression of the recombinant enzymes was still very low relative to the endogenous enzyme. Since we do not have a way to turn off the endogenous SAP genes while simultaneously turning on the pLexsy-sat2 containing genes, this pLexsy-sat2 expression system does not work for expression and subsequent isolation of our target enzymes. Future work will include investigating other expression systems to increase the yield of the recombinant enzyme relative to the endogenous SAPs.
Supplementary Material
References
- Vannier-Stantos MA, Martiny A, de Souza W (2002) Cell biology of Leishmania spp.: Invading and evading. Curr Pharm Des 8: 297-318.
- Berman JD (2003) Current treatment approaches to Leishmaniasis. Curr Op Infect Dis 16: 397-401.
- http://www.cdc.gov/parasites/leishmaniasis
- Pathak R, Batra S (2009) Malaria and Leishmaniasis: Current status of chemotherapy, new leads and targets for drug discovery. Anti-Infective Agents in Medicinal Chemistry 8: 226-267.
- Turner TL, Nguyen VH, McLauchlan CC, et al. (2012) Inhibitory effects of decavanadate on several enzymes and Leishmania tarentolae. Journal of Inorganic Biochemistry 108: 96-104.
- Gresser MJ, Tracy AS, Stankiewicz PJ (1987) The interaction of vanadate with tyrosine kinases and phosphatases. Advances in Protein Phosphatases 4: 35-37.
- Vannier Santos MA, Martiny A, Meyer-Fernandes JR, et al. (1995) Leishmanial protein kinase C modulates host cell interaction via secreted acid phosphatase. Eur J Cell Biol 67: 112-119.
- Mendez RS, Dorsey BM, McLauchlan CC, et al. (2014) Vanadium Complexes are in vitro Inhibitors of Leishmania Secreted Acid Phosphatases. International Journal of Chemistry 6: 35-49
- Ilg T, Overath P, Ferguson MAJ, et al. (1994) O- and N-glycosylation of the Leishmania mexicana-secreted acid phosphatase. Characterization of a new class of posphoserine-linked glycans. J Biol Chem 39: 24073-24081.
- Gottlieb M, Dwyer DW (1982) Identification and partial characterization of an extracellular acid phosphatase activity of Leishmania donovani promastigotes. Mol Cell Biol 2: 76-81.
- Ilg T, Stierhof YD, Etges R, et al. (1991) Secreted acid phosphatase of Leishmania mexicana: a filamentous phosphoglycoprotein polymer. Proc Natl Acad Sci USA 88: 8774-8778.
Corresponding Author
Marjorie A Jones, Department of Chemistry, Illinois State University, Normal, IL 61790-4160, USA, Tel: 1-309-438-2366.
Copyright
© 2017 Baumhardt JM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.